404
- -Page not found
- - -Page not found
- - -Page not found
- - -![]()
nilearn and nibabelGoogle Colaboratory is is a free cloud-based platform provided by Google that offers a Jupyter notebook environment for writing and executing Python code. It is primarily used for data analysis tasks but can also be used for general-purpose Python programming. It has many benefits when working with data (including neuroimaging data) such as:
-It is an interactive environment similar to Anaconda, but with certain advantages (like those mentioned above). Similarly, Colab allows for users to run code in small chunks called 'cells', displaying any output such as images directly within the notebook as well. The programming language used by these notebooks is Python, which it organises in the form of Colab Notebooks.
What can we do in Google Colab? We can do a whole bunch of things...
-import matplotlib.pyplot as plt
-import numpy as np
-
-x = np.linspace(0, 10, 100)
-y = np.sin(x)
-
-plt.plot(x, y)
-plt.title("Sine Wave")
-plt.xlabel("X")
-plt.ylabel("sin(X)")
-plt.show()
-
import numpy as np
-import matplotlib.pyplot as plt
-
-def mandelbrot(c, max_iter):
- z = 0
- n = 0
- while abs(z) <= 2 and n < max_iter:
- z = z*z + c
- n += 1
- return n
-
-def mandelbrot_image(xmin, xmax, ymin, ymax, width=10, height=10, max_iter=256):
- # Create a width x height grid of complex numbers
- x = np.linspace(xmin, xmax, width * 100)
- y = np.linspace(ymin, ymax, height * 100)
- X, Y = np.meshgrid(x, y)
- C = X + 1j * Y
-
- # Compute Mandelbrot set
- img = np.zeros(C.shape, dtype=int)
- for i in range(width * 100):
- for j in range(height * 100):
- img[j, i] = mandelbrot(C[j, i], max_iter)
-
- # Plotting
- plt.figure(figsize=(width, height))
- plt.imshow(img, extent=(xmin, xmax, ymin, ymax), cmap='hot')
- plt.colorbar()
- plt.title("Mandelbrot Set")
- plt.show()
-
-# Parameters defining the extent of the region in the complex plane we're going to plot
-xmin, xmax, ymin, ymax = -2.0, 0.5, -1.25, 1.25
-width, height = 10, 10
-max_iter = 256
-
-mandelbrot_image(xmin, xmax, ymin, ymax, width, height, max_iter)
-
from IPython.display import YouTubeVideo
-
-# Embedding the YouTube video
-YouTubeVideo('GtL1huin9EE', width=800, height=450)
--
-![]()
nibabel is designed to provide a simple, uniform interface to various neuroimaging file formats, allowing users to easily manipulate neuroimaging data within Python scripts or interactive environments like Jupyter notebooks.
Key features and capabilities of nibabel include:
Reading and Writing Neuroimaging Data: nibabel allows you to read data from disk into Python data structures and write data back to disk in various neuroimaging formats.
Data Manipulation: Once loaded into Python, neuroimaging data can be manipulated just like any other data structure. This includes operations like slicing, statistical analyses, and visualization.
-Header Access: nibabel provides access to the headers of neuroimaging files, which contain metadata about the imaging data such as dimensions, voxel sizes, data type, and orientation. This is crucial for understanding and correctly interpreting the data.
Affine Transformations: It supports affine transformations that describe the relationship between voxel coordinates and world coordinates, enabling spatial operations on the data.
-nilearn is a Python library designed to facilitate fast and easy statistical learning analysis and manipulation of neuroimaging data. It builds on libraries such as numpy, scipy, scikit-learn, and pandas, offering a comprehensive toolkit for neuroimaging data processing, with a focus on machine learning applications. nilearn aims to make it easier for researchers in neuroscience and machine learning to use Python for sophisticated imaging data analysis and visualization.
In this intro, we won't be focusing on the more advanced uses such as those involving machine learning, but just leveraging it's ability to perform simple functions with structural MRI images.
-To this end, one of the strengths of nilearn is its powerful and flexible plotting capabilities, designed specifically for neuroimaging data. It provides functions to visualize MRI volumes, statistical maps, connectome diagrams, and more, with minimal code.
Before we get started, we need to install the necessary packages and import the NIFTIs. We will use T1-weighted structural MRI scans, one of myself and one that I copied from OpenNeuro, a website for openly available neuroimaging datasets.
-# Install necessary packages
-%%capture
-!pip install nibabel matplotlib nilearn
-!pip install imageio
-
-# Download the MRI scan files from GitHub and rename them
-!wget https://raw.githubusercontent.com/sohaamir/MRICN/main/niftis/chris_T1.nii -O chris_brain.nii
-!wget https://raw.githubusercontent.com/sohaamir/MRICN/main/niftis/aamir_T1.nii -O aamir_brain.nii
-
-import nibabel as nib
-import nilearn as nil
-import numpy as np
-import pylab as plt
-import matplotlib.pyplot as plt
-import imageio
-import os
-nibabel?The code below loads a NIfTI image and prints out its shape and data type. The shape tells us the dimensions of the image, which typically includes the number of voxels in each spatial dimension (X, Y, Z) and sometimes time or other dimensions. The data type indicates the type of data used to store voxel values, such as float or integer types.
-# Load the first image using nibabel
-t1_aamir_path = 'aamir_brain.nii'
-t1_aamir_image = nib.load(t1_aamir_path)
-
-# Now let's check the image data shape and type
-print("Aamir's image shape:", t1_aamir_image.shape)
-print("Aamir's image data type:", t1_aamir_image.get_data_dtype())
-Aamir's image shape: (192, 256, 256)
-Aamir's image data type: int16
-Each NIfTI file also comes with a header containing metadata about the image. Here we've extracted the voxel sizes, which represent the physical space covered by each voxel, and the image orientation, which tells us how the image data is oriented in space.
-# Access the image header
-header_aamir = t1_aamir_image.header
-
-# Print some header information
-print("Aamir's voxel size:", header_aamir.get_zooms())
-print("Aamir's image orientation:", nib.aff2axcodes(t1_aamir_image.affine))
-Aamir's voxel size: (0.94, 0.9375, 0.9375)
-Aamir's image orientation: ('R', 'A', 'S')
-We can visualize the data from our NIfTI file by converting it to a numpy array and then using matplotlib to display a slice. In this example, we've displayed an axial slice from the middle of the brain, which is a common view for inspecting T1-weighted images.
# Get the image data as a numpy array
-aamir_data = t1_aamir_image.get_fdata()
-
-# Display one axial slice of the image
-aamir_slice_index = aamir_data.shape[1] // 2 # Get the middle index along Z-axis
-plt.imshow(aamir_data[:, :, aamir_slice_index], cmap='gray')
-plt.title('Middle Axial Slice of Aamirs Brain')
-plt.axis('off') # Hide the axis to better see the image
-plt.show()
-
Now we can load a second T1-weighted image and printed its shape for comparison. By comparing the shapes of the two images, we can determine if they are from the same scanning protocol or if they need to be co-registered for further analysis.
-# Load the second image
-t1_chris_path = 'chris_brain.nii'
-t1_chris_image = nib.load(t1_chris_path)
-
-# Let's compare the shapes of the two images
-print("First image shape:", t1_aamir_image.shape)
-print("Second image shape:", t1_chris_image.shape)
-First image shape: (192, 256, 256)
-Second image shape: (176, 240, 256)
-Now that we have loaded both T1-weighted MRI images, we are interested in visualizing and comparing them directly. To do this, we will extract the data from the second image, chris_brain.nii, and display a slice from it alongside a slice from the first image, aamir_brain.nii.
# Get data for the second image
-chris_data = t1_chris_image.get_fdata()
-chris_slice_index = chris_data.shape[1] // 2 # Get the middle index along Z-axis
-
-# Display a slice from both images side by side
-fig, axes = plt.subplots(1, 2, figsize=(12, 6))
-
-# Plot first image slice
-axes[0].imshow(aamir_data[:, :, aamir_slice_index], cmap='gray')
-axes[0].set_title('aamir_brain.nii')
-axes[0].axis('off')
-
-# Plot second image slice
-axes[1].imshow(chris_data[:, :, chris_slice_index], cmap='gray')
-axes[1].set_title('chris_brain.nii')
-axes[1].axis('off')
-
-plt.show()
-
To visualize the structure of the brain in MRI scans, we can create an animated GIF that scrolls through each slice of the scan. This is particularly useful for examining the scan in a pseudo-3D view by observing one slice at a time through the entire depth of the brain.
-The following code defines a function create_gif_from_mri_normalized that processes an MRI scan file and produces an animated GIF. The MRI data is first normalized by clipping the top and bottom 1% of pixel intensities, which enhances contrast and detail. The scan is then sliced along the sagittal plane, and each slice is converted to an 8-bit grayscale image and compiled into an animated GIF. This normalization process ensures that the resulting GIF maintains visual consistency across different scans.
We apply this function to two MRI scans, aamir_brain.nii and chris_brain.nii, creating a GIF for each. These GIFs, named 'aamir_brain_normalized.gif' and 'chris_brain_normalized.gif', respectively, will allow us to visually assess and compare the scans.
# Function to normalize and create a GIF from a 3D MRI scan in the sagittal plane
-def create_gif_from_mri_normalized(path, gif_name):
- # Load the image and get the data
- img = nib.load(path)
- data = img.get_fdata()
-
- # Normalize the data for better visualization
- # Clip the top and bottom 1% of pixel intensities
- p2, p98 = np.percentile(data, (2, 98))
- data = np.clip(data, p2, p98)
- data = (data - np.min(data)) / (np.max(data) - np.min(data))
-
- # Prepare to capture the slices
- slices = []
- # Sagittal slices are along the x-axis, hence data[x, :, :]
- for i in range(data.shape[0]):
- slice = data[i, :, :]
- slice = np.rot90(slice) # Rotate or flip the slice if necessary
- slices.append((slice * 255).astype(np.uint8)) # Convert to uint8 for GIF
-
- # Create a GIF
- imageio.mimsave(gif_name, slices, duration=0.1) # duration controls the speed of the GIF
-
-# Create GIFs from the MRI scans
-create_gif_from_mri_normalized('aamir_brain.nii', 'aamir_brain_normalized.gif')
-create_gif_from_mri_normalized('chris_brain.nii', 'chris_brain_normalized.gif')
-
-After generating the GIFs for each MRI scan, we can now display them directly within the notebook. This visualization provides us with an interactive view of the scans, making it easier to observe the entire brain volume as a continuous animation.
-Below, we use the IPython.display module to render the GIFs in the notebook. The first GIF corresponds to Aamir's brain scan, and the second GIF corresponds to Chris's brain scan. These inline animations can be a powerful tool for presentations, education, and qualitative analysis, offering a dynamic view into the MRI data without the need for specialized neuroimaging software.
from IPython.display import Image, display
-
-# Display the GIF for Aamir's MRI
-display(Image(filename='aamir_brain_normalized.gif'))
-
-# Display the GIF for Chris's MRI
-display(Image(filename='chris_brain_normalized.gif'))
-<IPython.core.display.Image object>
-
-
-
-<IPython.core.display.Image object>
-nilearn?With nilearn, we can create informative visualizations of brain images. One common technique is to overlay a statistical map or a labeled atlas on top of an anatomical image for better spatial context. Here, we will demonstrate how to overlay a standard atlas on our T1-weighted MRI images. This allows us to see how different brain regions delineated by the atlas correspond to structures within the actual brain images.
from nilearn import plotting, datasets
-
-# Load the atlas
-atlas_data = datasets.fetch_atlas_harvard_oxford('cort-maxprob-thr25-2mm')
-
-# Plotting the atlas map overlay on the first image
-plotting.plot_roi(atlas_data.maps, bg_img='aamir_brain.nii', title="Aamir's Brain with Atlas Overlay", display_mode='ortho', cut_coords=(0, 0, 0), cmap='Paired')
-
-# Plotting the atlas map overlay on the second image
-plotting.plot_roi(atlas_data.maps, bg_img='chris_brain.nii', title="Chris's Brain with Atlas Overlay", display_mode='ortho', cut_coords=(0, 0, 0), cmap='Paired')
-
-plotting.show()
-Added README.md to /root/nilearn_data
-
-
-Dataset created in /root/nilearn_data/fsl
-
-Downloading data from https://www.nitrc.org/frs/download.php/9902/HarvardOxford.tgz ...
-
-
- ...done. (1 seconds, 0 min)
-Extracting data from /root/nilearn_data/fsl/c4d84bbdf5c3325f23e304cdea1e9706/HarvardOxford.tgz..... done.
-

Note that the atlas is formatted correctly on Chris's brain but not Aamir's. Why do you think this is?
-nilearn also offers convenient functions for visualizing neuroimaging data. It can handle various brain imaging data formats and provides easy-to-use tools for plotting. Here, we will use nilearn to plot axial slices from our two T1-weighted MRI images, aamir_brain.nii and chris_brain.nii. This is a bit different to how we plotted the data before using nibabel.
from nilearn import plotting
-
-# Plotting the axial view of Aamir's brain
-aamir_img = 'aamir_brain.nii'
-plotting.plot_anat(aamir_img, title="Axial View of Aamir's Brain", display_mode='z', cut_coords=10)
-
-# Plotting the axial view of Chris's brain
-chris_img = 'chris_brain.nii'
-plotting.plot_anat(chris_img, title="Axial View of Chris's Brain", display_mode='z', cut_coords=10)
-
-plotting.show()
-

With nilearn, we can also perform statistical analysis on the brain imaging data. For instance, we can calculate and plot the mean intensity of the brain images across slices. This can reveal differences in signal intensity and distribution between the two scans, which might be indicative of varying scan parameters or anatomical differences.
import numpy as np
-from nilearn.image import mean_img
-
-# Calculating the mean image for both scans
-mean_aamir_img = mean_img(aamir_img)
-mean_chris_img = mean_img(chris_img)
-
-# Plotting the mean images
-plotting.plot_epi(mean_aamir_img, title="Mean Image of Aamir's Brain")
-plotting.plot_epi(mean_chris_img, title="Mean Image of Chris's Brain")
-
-plotting.show()
-

nilearn is not just for plotting—it also provides tools for image manipulation and transformation. We can apply operations such as smoothing, filtering, or masking to the brain images. Below, we will smooth the images using a Gaussian filter, which is often done to reduce noise and improve signal-to-noise ratio before further analysis.
Since this is usually performed just on the brain (i.e., not the entire image), let's smooth the extracted brain using FSL.
-!wget https://raw.githubusercontent.com/sohaamir/MRICN/main/niftis/chris_bet.nii -O chris_bet.nii
-!wget https://raw.githubusercontent.com/sohaamir/MRICN/main/niftis/aamir_bet.nii -O aamir_bet.nii
-
-from nilearn import image
-from nilearn.image import smooth_img
-
-# Load the skull-stripped brain images
-chris_img = image.load_img('chris_bet.nii')
-aamir_img = image.load_img('aamir_bet.nii')
-
-# Applying a Gaussian filter for smoothing (with 4mm FWHM)
-smoothed_aamir_img = smooth_img(aamir_img, fwhm=4)
-smoothed_chris_img = smooth_img(chris_img, fwhm=4)
-
-# Save the smoothed images to disk
-smoothed_aamir_img.to_filename('smoothed_aamir_brain.nii')
-smoothed_chris_img.to_filename('smoothed_chris_brain.nii')
-
-# Plotting the smoothed images
-plotting.plot_anat(smoothed_aamir_img, title="Smoothed Aamir's Brain")
-plotting.plot_anat(smoothed_chris_img, title="Smoothed Chris's Brain")
-
-plotting.show()
-

Meet Chris:
-He's a Senior Product Manager at Google. Before that he was Co-Director of the Center for Reproducible Neuroscience at Stanford University. Before that he was a postdoc at the Max Planck Institute for Human Cognitive and Brain Sciences, and before that he completed a PhD at the University of Edinburgh.
-Meet Aamir:
-He's a PhD student in Psychology at the University of Birmingham. Before that he worked as a Junior Research Fellow and Operations Support at the University of Reading. Before that he did an MSc in Brain Imaging at the University of Nottingham, and before that he completed a BSc in Biomedical Science at Imperial College London.
-Given these two profiles, how can we discern who is more intelligent?
-This is a question that has puzzled psychologists for way over a century, but let's take the opinion that you can generally who is more intelligent by the size of their brain.
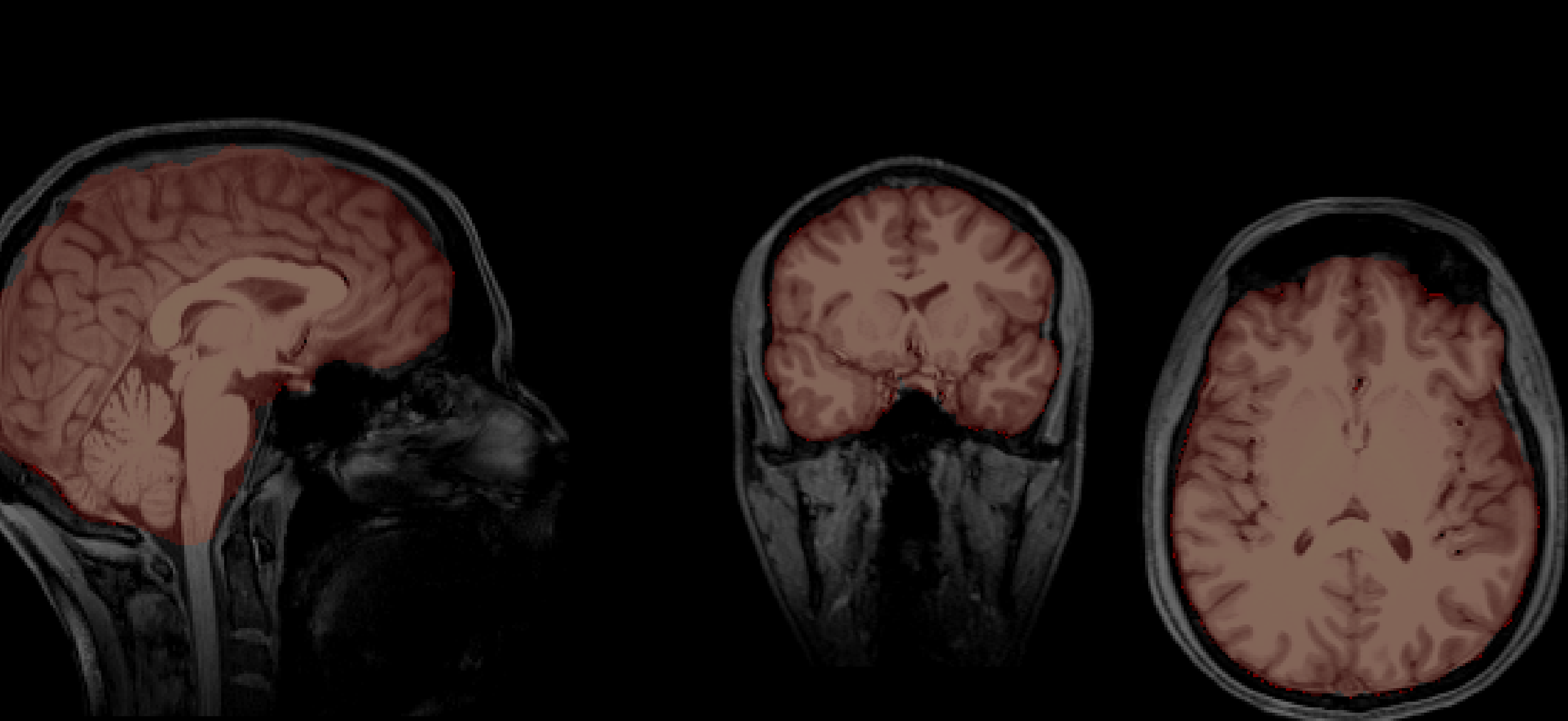
-Again, let's take the two brain images that I have skull-stripped using BET in FSL.
-Chris's brain
-
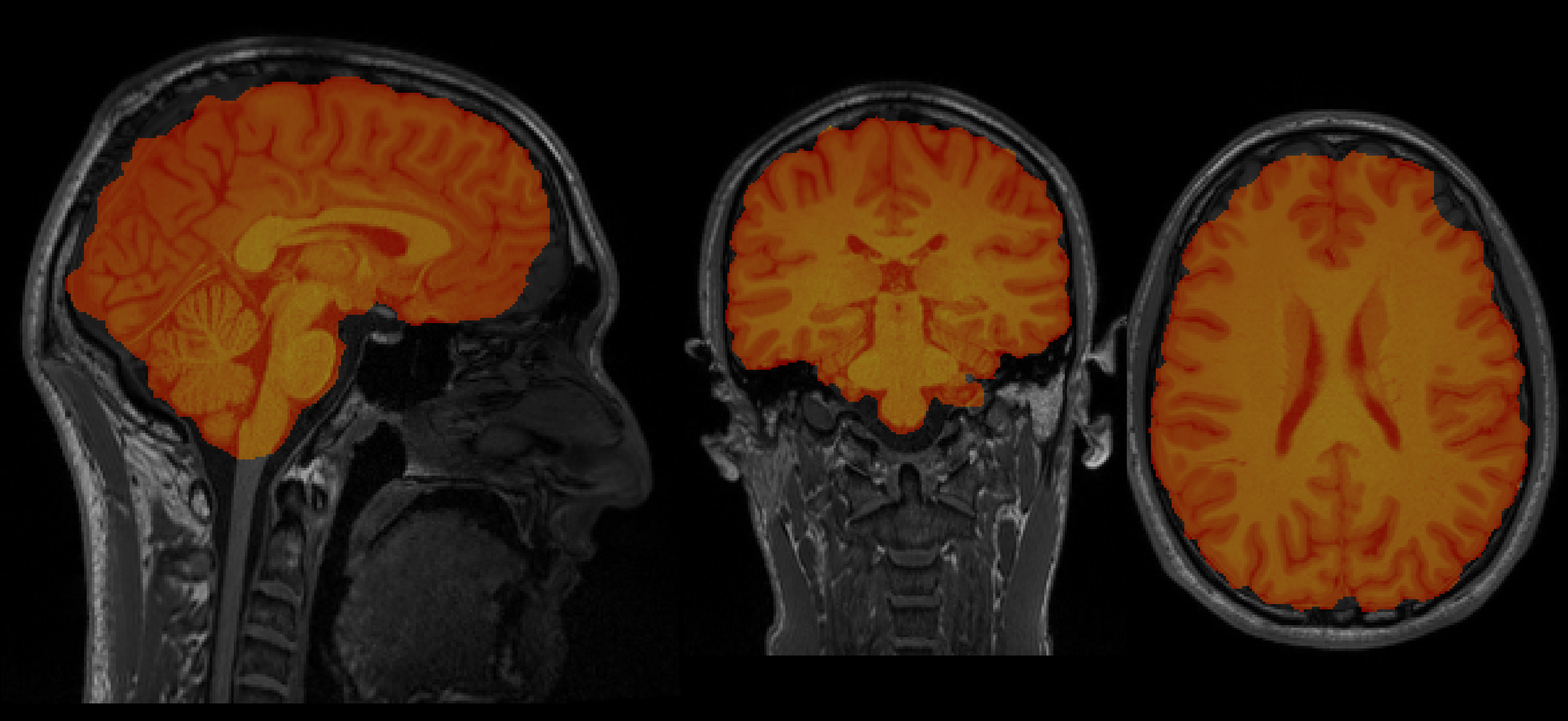
Aamir's brain
-
So now let's see who's brain is bigger (place your BETs!) -(pun intended)
-from nilearn import image
-
-# Convert the images to data arrays
-chris_data = chris_img.get_fdata()
-aamir_data = aamir_img.get_fdata()
-
-# Calculate the size by counting the non-zero voxels in the brain mask
-chris_size = np.sum(chris_data > 0)
-aamir_size = np.sum(aamir_data > 0)
-
-# Print out the sizes and determine which is larger
-print(f"Chris's brain size: {chris_size} voxels")
-print(f"Aamir's brain size: {aamir_size} voxels")
-
-# Determine which brain is larger
-if chris_size > aamir_size:
- print("Chris has the larger brain.")
-elif aamir_size > chris_size:
- print("Aamir has the larger brain.")
-else:
- print("Both brains are the same size.")
-Chris's brain size: 1480032 voxels
-Aamir's brain size: 1799361 voxels
-Aamir has the larger brain.
-After running this we get:
-Chris's brain size: 1480032 voxels
-Aamir's brain size: 1799361 voxels
-Aamir has the larger brain.
-So, it seems as if Aamir has the bigger brain. We can doublecheck these results by running the same analysis using fslstats.
The code to run this on your local machine is:
-# Calculate the number of non-zero voxels for Chris' brain
-chris_voxels=$(fslstats chris_bet.nii -V | awk '{print $1}')
-
-# Calculate the number of non-zero voxels for Aamir's brain
-aamir_voxels=$(fslstats aamir_bet.nii -V | awk '{print $1}')
-
-# Print the number of voxels
-echo "Chris' brain size in voxels: $chris_voxels"
-echo "Aamir's brain size in voxels: $aamir_voxels"
-
-# Compare the voxel counts and print who has the bigger brain
-if [ "$chris_voxels" -gt "$aamir_voxels" ]; then
- echo "Chris has the bigger brain."
-elif [ "$chris_voxels" -lt "$aamir_voxels" ]; then
- echo "Aamir has the bigger brain."
-else
- echo "Both brains are the same size."
-fi
-which gives:
-Chris' brain size in voxels: 1480032
-Aamir's brain size in voxels: 1799361
-Aamir has the bigger brain.
-It's a nice sanity check to see that both nilearn and FSL reach the same conclusion in Aamir having the bigger brain.
![]()
The brain if often described as a 'connectome' that is it's regions are connected to each other, both structurally and functionally. Diffusion MRI was a method by which you could measure the former, whilst functional connectivity is a way by which we can do the latter.
-Functional connectivity is a broad definition, and has many different statistical methodologies under it's umbrella. For the sake of this tutorial however, we will stick to the most rudimentary definition: a correlation between two time-series (voxel or region-wise) during a scan.
-In this notebook, we will walk through the basics of functional connectivity analysis, using an example dataset from the nilearn package.
Credits to Lucas Snoek's notebook on nilearn and the official nilearn guide to functional connectivity analysis.
Before we get started, let's load up some programming music...
-from IPython.display import YouTubeVideo
-
-# Embed the YouTube video
-YouTubeVideo('jfKfPfyJRdk')
-We will need to install/import a bunch of packages including:
-nilearn to work with our imaging datamatplotlib for plottingnumpy and scipy for mathsy stuff%load_ext autoreload
-%autoreload 2
-# Install necessary packages
-%%capture
-!pip install matplotlib nilearn
-
-import nilearn as nil
-import numpy as np
-import matplotlib.pyplot as plt
-import scipy
-From nilearn, we can import 'resting-state' fMRI data from the Richardson dataset where 'children and adults watched Disney Pixar’s “Partly Cloudy” while lying in the scanner. There was no task; participants were simply instructed to lie still and watch the movie'.
Movie-watching has recently become an alternative paradigm to conventional 'resting-state' paradigms, see (Finn & Bandettini, 2021) for more information.
-For the purposes of this notebook, it doesn't matter too much what data we have, so long as it's functional.
-Because this is just an example, we will just analyse a single participant's scan. In the code below, we fetch both a brain atlas (Harvard-Oxford) and a single subject's fMRI data. We then create a 'masker object' (i.e., a mask) using the atlas, and then apply the mask to extract the time series data from the fMRI data for al regions in the atlas while regressing out confounding variables.
-As the functional data is already registered to standard space, we load the Harvard-Oxford atlas and define voxels within the brain as exclusively belonging to different regions of the atlas.
-from nilearn import datasets
-from nilearn.maskers import NiftiLabelsMasker
-
-dataset = datasets.fetch_atlas_harvard_oxford("cort-maxprob-thr25-2mm")
-atlas_filename = dataset.maps
-labels = dataset.labels
-
-print(f"Atlas ROIs are located in nifti image (4D) at: {atlas_filename}")
-
-# One subject of brain development fMRI data
-data = datasets.fetch_development_fmri(n_subjects=1, reduce_confounds=True)
-fmri_filenames = data.func[0]
-reduced_confounds = data.confounds[0] # This is a preselected set of confounds
-
-masker = NiftiLabelsMasker(
- labels_img=atlas_filename,
- standardize="zscore_sample",
- standardize_confounds="zscore_sample",
- memory="nilearn_cache",
- verbose=5,
-)
-
-# Here we go from nifti files to the signal time series in a numpy array
-# We also give confounds to be regressed out during signal extraction
-time_series = masker.fit_transform(fmri_filenames, confounds=reduced_confounds)
-But we are interesting in correlating the time-series between regions. So, in order to do this we just take the mean of the time-series for each region.
-Specfically, in the code below we create a function to take all the voxels that belong to the same region (or label as it's defined in this instance) and calculate the mean activity across all voxels for each region in the scan.
-This is equivalent to performing functional connectivity using fslstats in FSL, where you might run the following:
fslmeants -i input/to/subject_data -o output.txt -m brain_region_mask
We've just done this for all regions in the Harvard-Oxford atlas in just a few lines of code.
-We can then plot the fMRI data with the atlas overlaid:
-from nilearn import plotting, image
-import matplotlib.pyplot as plt
-
-# Compute the mean fMRI image (across the time dimension)
-mean_fmri_img = image.mean_img(fmri_filenames)
-
-# Plot the mean fMRI image with the Harvard-Oxford atlas overlaid
-# Adjusting the figure size here
-fig, ax = plt.subplots(figsize=(8, 6)) # Smaller figure size
-display = plotting.plot_anat(mean_fmri_img, display_mode='ortho',
- title="Mean fMRI Image with Harvard-Oxford Atlas Overlay",
- cut_coords=(0, 0, 0), axes=ax)
-display.add_overlay(atlas_filename, cmap=plotting.cm.bwr, alpha=0.5)
-
-# Show plot
-plt.show()
-
Let's plot the mean time-series for each region to show this.
-import matplotlib.pyplot as plt
-
-# Determine the number of unique regions
-num_regions = len(labels) - 1
-
-# Set up the figure size
-plt.figure(figsize=(15, 5)) # Adjusted for better visualization in a single plot
-
-# Plot the time series for each region on the same plot
-for i in range(1, num_regions + 1):
- plt.plot(time_series[:, i - 1], label=labels[i])
-
-plt.title('Time Series for All Regions')
-plt.xlabel('Time Points')
-plt.ylabel('Signal')
-plt.tight_layout()
-
-plt.show()
-
So, since our definition of functional connectivity is the degree of similarity between two timeseries, we can see regions that have a high and low connectivity by first identifying them by running a correlation across all regions and printing out which two regions have the highest and lowest:
-# Find the indices of the maximum and minimum value in the correlation matrix
-max_corr = np.unravel_index(np.argmax(np.abs(correlation_matrix), axis=None), correlation_matrix.shape)
-min_corr = np.unravel_index(np.argmin(np.abs(correlation_matrix[np.nonzero(correlation_matrix)])), correlation_matrix.shape)
-
-# Extract indices for strongest and weakest connections
-region1, region2 = max_corr
-region1_weak, region2_weak = min_corr
-
-print(f"The two regions with the strongest connectivity are {labels[region1]} and {labels[region2]}, with a correlation of {correlation_matrix[region1, region2]:.2f}.")
-print(f"The two regions with the weakest connectivity are {labels[region1_weak]} and {labels[region2_weak]}, with a correlation of {correlation_matrix[region1_weak, region2_weak]:.2f}.")
-The two regions with the strongest connectivity are Subcallosal Cortex and Paracingulate Gyrus, with a correlation of 0.81.
-The two regions with the weakest connectivity are Superior Parietal Lobule and Superior Temporal Gyrus, posterior division, with a correlation of -0.02.
-We can plot the mean connectivity across the region at each time-point. Remember that the degree of functional connectivity is most simply assessed by calculating the correlation coefficient between the time series of different brain regions. A high correlation coefficient (close to +1 or -1) indicates strong functional connectivity, while a coefficient close to 0 indicates weak or no functional connectivity.
-import matplotlib.pyplot as plt
-import numpy as np
-from scipy import stats
-
-# Function to plot scatter graph with line of best fit and R value, with unique color for each time point
-def plot_scatter_with_fit(x, y, labels, ax, title, line_color):
- # Calculate the line of best fit
- slope, intercept, r_value, p_value, std_err = stats.linregress(x, y)
- line = slope * x + intercept
-
- # Generate a color map based on the length of x or y
- colors = plt.cm.jet(np.linspace(0, 1, len(x)))
-
- # Plot the scatter graph with a unique color for each time point
- for i in range(len(x)):
- ax.scatter(x[i], y[i], alpha=0.5, color=colors[i])
- ax.plot(x, line, color=line_color, label=f'Fit: R={r_value:.2f}')
- ax.set_xlabel(f"{labels[0]} Signal")
- ax.set_ylabel(f"{labels[1]} Signal")
- ax.set_title(title)
- ax.legend()
-
-# Assuming time_series, labels, region1, region2, region1_weak, region2_weak are defined
-time_series_region1 = time_series[:, region1]
-time_series_region2 = time_series[:, region2]
-time_series_region1_weak = time_series[:, region1_weak]
-time_series_region2_weak = time_series[:, region2_weak]
-
-# Set up a figure with two subplots side by side for scatter plots
-fig, axes = plt.subplots(1, 2, figsize=(14, 6))
-
-line_color = 'red' # Color for the line of best fit
-
-# Scatter plot for the two strongly connected regions with unique color for each time point
-plot_scatter_with_fit(time_series_region1, time_series_region2,
- [f"Region {labels[region1]}", f"Region {labels[region2]}"],
- axes[0], 'Strongly Connected Regions',
- line_color)
-
-# Scatter plot for the two weakly connected regions with unique color for each time point
-plot_scatter_with_fit(time_series_region1_weak, time_series_region2_weak,
- [f"Weak Region {labels[region1_weak]}", f"Weak Region {labels[region2_weak]}"],
- axes[1], 'Weakly Connected Regions',
- line_color)
-
-plt.tight_layout()
-plt.show()
-
-
Each dot represents one TR in the time-series. The similarity is measured by the simiality between the signal in the same TRs across both regions.
-To see this more clearly, we can plot the mean time-series for each region.
-import matplotlib.pyplot as plt
-import numpy as np
-
-# Assuming time_series, labels, region1, region2, region1_weak, region2_weak are defined from previous steps
-
-# Extract the time series for the regions
-time_series_region1 = time_series[:, region1]
-time_series_region2 = time_series[:, region2]
-time_series_region1_weak = time_series[:, region1_weak]
-time_series_region2_weak = time_series[:, region2_weak]
-
-# Set up a figure with two subplots side by side for time series
-fig, axes = plt.subplots(1, 2, figsize=(15, 5))
-
-# Plot the time series for the two strongly connected regions on the first subplot
-axes[0].plot(time_series_region1, label=f"Region {labels[region1]}")
-axes[0].plot(time_series_region2, label=f"Region {labels[region2]}")
-axes[0].set_xlabel('Time points')
-axes[0].set_ylabel('Signal')
-axes[0].set_title('Time Series for the Two Strongly Connected Regions')
-axes[0].legend()
-
-# Plot the time series for the two weakly connected regions on the second subplot
-axes[1].plot(time_series_region1_weak, label=f"Weak Region {labels[region1_weak]}")
-axes[1].plot(time_series_region2_weak, label=f"Weak Region {labels[region2_weak]}")
-axes[1].set_xlabel('Time points')
-axes[1].set_ylabel('Signal')
-axes[1].set_title('Time Series for the Two Weakly Connected Regions')
-axes[1].legend()
-
-plt.tight_layout()
-plt.show()
-
Functional connectivity analysis in this manner is correlational not causal, so we should be aware of that. However, for the purposes of our tutorial, this is just fine, and it is actually the same approach that the workshop tutorial uses.
-So, let's give that a go!
- -This tutorial is based upon a previous tutorial: FSL fMRI Resting State Seed-based Connectivity by Dianne Paterson at the University of Arizona, but which has been -adapted to run on the BEAR systems at the University of Birmingham, and has some -additional material added.
-In this session, we will run a group-level functional connectivity analysis on -resting-state fMRI data of three participants. We will specifically look at the -functional connectivity of the posterior cingulate cortex (PCC), a region of the -default mode network (DMN) that is commonly found to be active in resting-state data.
-To do this, we will:
-There hopefully shouldn't be any issues, but pelase let me know if there are!
-Navigate to your shared directory within the MRICN folder and copy the data:
-cd /rds/projects/c/chechlmy-chbh-mricn/xxx
-cp -r /rds/projects/c/chechlmy-chbh-mricn/aamir_test/SBC .
-cd SBC
-ls
-You should see the following:
-sub1 sub2 sub3
-Each of the folders has a single resting-state scan, called sub1.nii.gz,
-sub2.nii.gz and sub3.nii.gz respectively.
We will now create our seed region for the PCC. To do this, firstly load FSL and -fsleyes in the terminal by running:
-module load FSL/6.0.5.1-foss-2021a
-module load FSLeyes/1.3.3-foss-2021a
-Check that we are in the correct directory (blah/your_username/SBC):
-pwd
-and create a new directory called seed:
mkdir seed
-Now when you run ls you should see:
seed sub1 sub2 sub3
-Lets open FSLeyes:
-fsleyes &
-We need to open the standard MNI template brain, select the PCC and make a mask.
-Here are the following steps:
-1) Navigate to the top menu and click on File ➜ Add standard and select MNI152_T1_2mm_brain.nii.gz.
-2) When the image is open, click on Settings ➜ Ortho View 1 ➜ Atlases.
-An atlas panel opens on the bottom section.
-3) Select Atlas information (if it already hasn't loaded).
-4) Ensure Harvard-Oxford Cortical Structural Atlas is selected.
-5) Go into 'Atlas search' and type cing in the search box. Check the Cingulate Gyrus, posterior division (lower right) so that it is overlaid on the standard brain. (The full name may be obscured, but you can always check which region you have loaded by looking at
-the panel on the bottom right).
 -
-
-
-At this point, your window should look something like this:
-
-
- -
-
-
-To save the seed, click the save symbol which is the first of three icons on the
-bottom left of the window.
-
-
-
The window that opens up should be your project SBC directory. Open into the
-seed folder and save your seed as PCC.
We now need to binarise the seed and to extract the mean timeseries. To do this, leaving -FSLeyes open, go into your terminal (you may have to press Enter if some text about -dc.DrawText is there) and type:
-cd seed
-fslmaths PCC -thr 0.1 -bin PCC_bin
-In FSLeyes now click File ➜ Add from file, and select PCC_bin to compare PCC.nii.gz
-(before binarization) and PCC_bin.nii.gz (after binarization). You should note that the
-signal values are all 1.0 for the binarized PCC.

You can now close FSLeyes.
-For each subject, you want to extract the average time series from the region defined by -the PCC mask. To calculate this value for sub1, do the following:
-cd ../sub1
-fslmeants -i sub1 -o sub1_PCC.txt -m ../seed/PCC_bin
-This will generate a file within the sub1 folder called sub1_PCC.txt. We can
-have a look at the contents by running cat sub1_PCC.txt.The terminal will print
-out a bunch of numbers with the last five being:
20014.25528
-20014.919
-20010.17317
-20030.02886
-20066.05141
-This is the mean level of 'activity' for the PCC at each time-point.
-Now let's repeat this for the other two subjects.
-cd ../sub2
-fslmeants -i sub2 -o sub2_PCC.txt -m ../seed/PCC_bin
-cd ../sub3
-fslmeants -i sub3 -o sub3_PCC.txt -m ../seed/PCC_bin
-Now if you go back to the SBC directory and list all of the files within the -subject folders:
-cd ..
-ls -R
-You should see the following:
-
Please let me know if you do not get this!!
-This is all we need to run the subject and group-level analyses using FEAT.
-Close your terminal, open another one, move to your SBC folder, load FSL and open Feat:
-cd /rds/projects/c/chechlmy-chbh-mricn/xxx/SBC
-module load bear-apps/2022b
-module load FSL/6.0.7.6
-source $FSLDIR/etc/fslconf/fsl.sh
-Feat &
-We will run the first-level analysis for sub1. Set-up the following settings in -the respective tabs:
-Data
-Select 4D data: Click the folder icon, go into the sub1 folder and choose
-sub1.nii.gz`. Click OK. You will see a box saying that the 'Input file has a TR
-of 1...' this is fine, just click OK again.
Output directory: Click into the sub1 folder and click OK. Nothing will be in
-the right hand column, but that is because there are no folders within sub1. We
-will create our .feat folder within sub1.
This is what your data tab should look like (with the input data opened for show).
-
Pre-stats
-The data has already been pre-processed, so set Motion correction to 'None' and -uncheck BET. Your pre-stats should look like this:
-
Registration
-Nothing needs to be changed here.
-Stats
-Click on 'Full Model Setup' and do the following:
-1) Keep the Number of original EVs as 1.
-2) Type PCC for the EV name.
-3) Select Custom (1 entry per volume) for the Basic shape. Click into the sub1
-folder and select sub1_PCC.txt. This is the mean time series of the PCC for
-sub-001 and is the statistical regressor in our GLM model. This is different from
-analyses of task-based data which will usually have an events.tsv file with the
-onset times for each regressor of interest.
The first-level analysis will identify brain voxels that show a significant correlation -with the seed (PCC) time series data.
-4) Select 'None' for Convolution, and uncheck both 'Add temporal derivate' and -'Apply temporal filtering'.
-Your window should look like this:
-
In the same General Linear Model window, click the Contrast & F-tests tab, type -PCC in the title, and click Done. A blue and red design matrix is displayed. -You can close it.
-Post-stats
-Nothing needs to be changed here.
-You are ready to run the first-level analysis. Click Go to run. On BEAR, this -should only take a few minutes.
-To actually examine the output, go to the BEAR Portal and at the menu bar select
-Files ➜ /rds/projects/c/chechlmy-chbh-mricn/
-
-
- -
-
-
-Then go into SBC/sub1.feat, select report.html and click View (top left of
-the window). Navigate to the Post-stats tab and examine the outputs. It should
-look like this:
-
-
-
We can now run the second and third subjects. As we only have three subjects, we -could manually run the other two by just changing three things:
-1) The fMRI data path
-2) The output directory
-3) The sub_PCC.txt path
Whilst it would probably be quicker to do it manually in this case, it is not -practical in other instances (e.g., more subjects, subjects with different -number of scans etc.).. So, instead we will be scripting the first level FEAT -analyses for the other two subjects.
-(Go back into your terminal, you don't need to open a new terminal or close Feat)
-The setup for each analysis is saved as a specific file the design.fsf file
-within the FEAT output directory. We can see by opening the design.fsf file
-for sub1:
pwd # make sure you are in your SBC directory e.g., blah/xxx/SBC
-cd sub1.feat
-cat design.fsf
-Feat acts as a large 'function' with its many variables corresponding to the
-options that we choose when setting up in the GUI. We just need to change three
-of these (the three mentioned above) which in the design.fsf file correspond to:
set fmri(outputdir) "/rds/projects/c/chechlmy-chbh-mricn/xxx/SBC/sub1"
-set feat_files(1) "/rds/projects/c/chechlmy-chbh-mricn/xxx/SBC/sub1/sub1/"
-set fmri(custom1) "/rds/projects/c/chechlmy-chbh-mricn/xxx/SBC/sub1/sub1_PCC.txt"
-To do this, please copy the run_feat.sh script into your own SBC directory:
cd ..
-pwd # make sure you are in your SBC directory
-cp /rds/projects/c/chechlmy-chbh-mricn/axs2210/SBC/run_feat.sh .
-This is the script:
-#!/bin/bash
-
-# Prompt the user for the University account name
-read -p "Please enter your University account name: " account_name
-
-# Define the base directory with the user-provided account name
-base_dir="/rds/projects/c/chechlmy-chbh-mricn/${account_name}/SBC"
-
-echo "Using base directory: $base_dir"
-
-# Loop over each subject's data
-for sub in sub2 sub3; do
- # Define the input .nii.gz file for the subject
- input_file="${base_dir}/${sub}/${sub}.nii.gz"
-
- # Define the output FEAT directory
- output_dir="${base_dir}/${sub}.feat"
-
- # Define the custom EV file for the subject
- custom_ev_file="${base_dir}/${sub}/${sub}_PCC.txt"
-
- # Define the .fsf file for the subject
- design_file="${base_dir}/${sub}.fsf"
-
- # Copy the template design file from sub1 and modify it for the current subject
- cp "${base_dir}/sub1.feat/design.fsf" "$design_file"
-
- # Replace the input file path in the design file
- sed -i "s|set feat_files(1) \".*\"|set feat_files(1) \"${input_file}\"|g" "$design_file"
-
- # Replace the output FEAT directory in the design file
- sed -i "s|set fmri(outputdir) \".*\"|set fmri(outputdir) \"${output_dir}\"|g" "$design_file"
-
- # Replace the custom EV file in the design file
- sed -i "s|set fmri(custom1) \".*\"|set fmri(custom1) \"${custom_ev_file}\"|g" "$design_file"
-
- # Run FEAT analysis
- feat "$design_file"
-
- # Remove the .fsf file from the SBC directory after running FEAT
- rm -f "$design_file"
-done
-
-echo "FEAT analysis completed for sub2 and sub3."
-The first line is always needed to run bash scripts. The rest of the code just
-replaces the 3 things we wanted to change for the defined subs, sub2 and sub3.
You can have a look at the script yourself by typing cat run_bash.sh.
Run the code (from your SBC directory) by typing bash run_feat.sh
It will ask you for your University account name, this is your ADF username -(axs2210 for me).
-This should take about 5-10 minutes to run. In the meantime feel free to follow me -on GitHub @sohaamir and on Twitter @AamirNSohail (not part of the module but always -appreciated 😊).
-After they have finished running, if you have a look at the report.html file for both directories,
-they should look like this:
sub2
- -
-
-sub3
-
-
- -
-
-
Ok, so now we have our FEAT directories for all three subjects, we can run the
-group level analysis. Close Feat and open a new Feat by running Feat & in your
-SBC directory. Here is the group-level FEAT setup:
Data tab
-1) Change 'First-level analysis' to 'Higher-level analysis' -2) Keep the default option, for 'Inputs are lower-level FEAT directories'. -3) Keep the 'Number of inputs' as 3. -4) Click the 'Select FEAT directories'. Click the yellow folder on the right to select the FEAT folder that you had generated from each first-level analysis.
-By now, your window should look like this (before closing the Input window):
- -
-
-
-5) Keep 'Use lower-level copes ticked'.
-6) In 'Output directory' stay in your current directory (it should be SBC), and in the
-bottom bar, type in PCC_group at the end of the file path. Don't worry about it
-being empty, FSL will fill out the file path for us. If you click the folder again,
-this is what mine looks like:
 -
-
-Stats
Leave the 'Mixed effects: FLAME 1' and click Full model setup. -In the General Linear Model window, name the model 'PCC' and make sure the EVs are all 1s. The interface should look like this:
-
After that, click 'Done' and close the GLM design matrix that pops up. -(You don't need to change anything in the 'Contrasts and F-tests' tab).
-Post-stats
-Change the Z-threshold from 3.1 to 2.3. This is because we only have data from -3 subjects, and so aren't likely to see much at a high statistical threshold. If -running analysis on a larger group, we would usually keep this at 3.1.
-Click 'Go' to run!
-This should only take about 2-3 minutes. When this is running, you can load the
-report.html through the file browser as you did for the individual subjects. Click on
-the 'Results' tab, and then on 'Lower-level contrast 1 (PCC)'. Your results should
-look like this:

These are voxels demonstrating significant functional connectivity with the PCC -at a group-level (Z > 2.3).
-So, we have just ran our group-level analysis. Let's have a closer look at the
-outputted data. Close FEAT and your terminal, open a new one, go to your SBC directory
-and load up FSL and FSLeyes:
cd /rds/projects/c/chechlmy-chbh-mricn/xxx/SBC
-module load FSL/6.0.5.1-foss-2021a
-module load FSLeyes/1.3.3-foss-2021a
-fsleyes &
-In fsleyes, open up the standard brain (Navigate to the top menu and click on 'File ➜
-Add standard' and select MNI152_T1_2mm_brain.nii.gz). Then add in our
-contrast image (File ➜ Add from file, and then go into the PCC_group.gfeat and
-then into cope1.feat and open the file thresh_zstat1.nii.gz).
When that is opened, change the colour to 'Red-Yellow' and the 'Minimum' up to -2.3 (The max should be around 3.12). If you set the voxel location to (42, 39, 52) -your screen should look like this:
- -
-
-
-This is the map that we saw in the report.html file. In fact we can double
-check this by changing the voxel co-ordinates to (45, 38, 46). Our image matches
-the one on the far right below:
Our thresholded image in fsleyes
-
-
- -
-
-
-The FEAT output
-
-
-
So we know which voxels demonstrate significant correlation with the PCC, but -what regions of the brain are they in? Let's go through two ways in which we can -work this out.
-Firstly, we can simply just overlap an atlas on the image and see which regions the -activated voxels fall under.
-To do this, navigate to the top menu and click on 'Settings ➜ Ortho View 1 ➜ Atlases'. -Then at the bottom middle of the window, select the 'Harvard-Oxford Cortical Structural -Atlas' and on the window directly next to it on the right, click 'Show/Hide'. The atlas -should have loaded up but is blocking the voxels. Change the Opacity to about a -quarter.
-
By having a look at the 'Location' window (bottom left) we can now see that significant voxels of activity are mainly found in the:
-Right superior lateral occipital cortex
-
Posterior cingulate cortex (PCC) / precuneus
- -
-
- -
-
-
-Alternatively, we can also use Neurosynth, a website where you
-can get the resting-statefunctional connectivity of any voxel location or brain region.
-It does this by extracting data from studies and performing a meta-analysis on brain
-imaging studies that have results associated with your voxel/region of interest.
If you click the following link, you will see regions demonstrating significant connectivity with the posterior cingulate.
-If you type (46, -70, 32) as co-ordinates in Neurosynth, and then into the MNI
-co-ordinates section in fsleyes, not into the voxel location, because Neurosynth works with MNI space, you can see that in both cases the right superior lateral occipital
-cortex is activated. Note that the orientations of left and right are different between
-Neurosynth and fslsyes.
This is a great result given that we only have three subjects!
-Neurosynth
- -
-
-
-FSLeyes
-
-
- -
-
-Congrats! You have ran (I imagine) your first functional connectivity analysis!
And that is also the end of the MRICN module 😢
- -
-
--Created by Aamir Sohail, University of Birmingham, for the Magnetic -Resonance Imaging in Cognitive Neuroscience (MRICN) module, Spring Term 2024.
-
These are the materials for my input to the MRICN module at the University of Birmingham, which consist of three main parts:
-nilearn and nibabel (not necessary at all but there if you want to learn).Here is a quick description of all three:
-![]() -
-
The aamir_vs_chris.ipynb is a Google Colab notebook demonstrating the basic functions of two Python packages, nibabel and nilearn for manipulating and plotting neuroimaging data. Nibabel is a Python library primarily used for reading and writing different types of neuroimaging data files, while Nilearn is a Python module used for conducting a wide variety of statistical analyses on neuroimaging data.
It is not necessary as part of the module to know this, and we are only scratching the surface of what nibabel and nilearn can do, but it presents an alternative approach to the conventional GUI-based alternative of FSL/FSLeyes.
Specifically, we go through the following uses:
-We then can practically demonstrate a simple case of statistical analysis by comparing the size of two brains, my own and Chris Gorgolewski's, a prime contributor to many open-source initiatives in neuroimaging, who's structural T1 scan is made publicly available through OpenNeuro.
-Ultimately, we do this both using nilearn and FSL and end up with the exact same result regardless!
Code for running the voxel comparison test in FSL:
-# Calculate the number of non-zero voxels for Chris' brain
-chris_voxels=$(fslstats chris_bet.nii -V | awk '{print $1}')
-
-# Calculate the number of non-zero voxels for Aamir's brain
-aamir_voxels=$(fslstats aamir_bet.nii -V | awk '{print $1}')
-
-# Print the number of voxels
-echo "Chris' brain size in voxels: $chris_voxels"
-echo "Aamir's brain size in voxels: $aamir_voxels"
-
-# Compare the voxel counts and print who has the bigger brain
-if [ "$chris_voxels" -gt "$aamir_voxels" ]; then
- echo "Chris has the bigger brain."
-elif [ "$chris_voxels" -lt "$aamir_voxels" ]; then
- echo "Aamir has the bigger brain."
-else
- echo "Both brains are the same size."
-fi
-![]()
This notebook interactively demonstrates the basis of functional connectivity, as we will define it in our analysis.
-Specifically, using nilearn, we can import 'resting-state' fMRI data from the Richardson dataset where 'children and adults watched Disney Pixar’s “Partly Cloudy” while lying in the scanner.
We then parcellate the regions, and take a very crude approach to functional connectivity by getting the mean time-series for each region, and then correlating that with all of the other regions in the brain. Subsequently, we can then plot the most and least-connected regions, as well as the individual time-series for the regions demonstrating the strongest and weakest correlation (or 'connectivity'):
-

The primary focus of today's workshop is to run a group-level functional connectivity analysis on resting-state fMRI data of three participants. We will specifically look at the functional connectivity of the posterior cingulate cortex (PCC), a region of the default mode network (DMN) that is commonly found to be active in resting-state data.
-To do this, we will:
-
 -
-' + escapeHtml(summary) +'
' + noResultsText + '
'); - } -} - -function doSearch () { - var query = document.getElementById('mkdocs-search-query').value; - if (query.length > min_search_length) { - if (!window.Worker) { - displayResults(search(query)); - } else { - searchWorker.postMessage({query: query}); - } - } else { - // Clear results for short queries - displayResults([]); - } -} - -function initSearch () { - var search_input = document.getElementById('mkdocs-search-query'); - if (search_input) { - search_input.addEventListener("keyup", doSearch); - } - var term = getSearchTermFromLocation(); - if (term) { - search_input.value = term; - doSearch(); - } -} - -function onWorkerMessage (e) { - if (e.data.allowSearch) { - initSearch(); - } else if (e.data.results) { - var results = e.data.results; - displayResults(results); - } else if (e.data.config) { - min_search_length = e.data.config.min_search_length-1; - } -} - -if (!window.Worker) { - console.log('Web Worker API not supported'); - // load index in main thread - $.getScript(joinUrl(base_url, "search/worker.js")).done(function () { - console.log('Loaded worker'); - init(); - window.postMessage = function (msg) { - onWorkerMessage({data: msg}); - }; - }).fail(function (jqxhr, settings, exception) { - console.error('Could not load worker.js'); - }); -} else { - // Wrap search in a web worker - var searchWorker = new Worker(joinUrl(base_url, "search/worker.js")); - searchWorker.postMessage({init: true}); - searchWorker.onmessage = onWorkerMessage; -} diff --git a/docs/search/search_index.json b/docs/search/search_index.json deleted file mode 100644 index 5e7ae5a..0000000 --- a/docs/search/search_index.json +++ /dev/null @@ -1 +0,0 @@ -{"config":{"indexing":"full","lang":["en"],"min_search_length":3,"prebuild_index":false,"separator":"[\\s\\-]+"},"docs":[{"location":"aamir_vs_chris/","text":"Who has the bigger brain, me or Chris Gorgolewski? A gentle introduction to manipulating neuroimaging data using Python with nilearn and nibabel What is Google Colab? Google Colaboratory is is a free cloud-based platform provided by Google that offers a Jupyter notebook environment for writing and executing Python code. It is primarily used for data analysis tasks but can also be used for general-purpose Python programming. It has many benefits when working with data (including neuroimaging data) such as: Free access Cloud-based hosting (available everywhere) GPU/TPU Support (lots of processing power) External Data Access (import data GitHub, Google Drive, local machine) It is an interactive environment similar to Anaconda, but with certain advantages (like those mentioned above). Similarly, Colab allows for users to run code in small chunks called 'cells', displaying any output such as images directly within the notebook as well. The programming language used by these notebooks is Python , which it organises in the form of Colab Notebooks . What can we do in Google Colab? We can do a whole bunch of things... Data visualization and plotting import matplotlib.pyplot as plt import numpy as np x = np.linspace(0, 10, 100) y = np.sin(x) plt.plot(x, y) plt.title(\"Sine Wave\") plt.xlabel(\"X\") plt.ylabel(\"sin(X)\") plt.show() import numpy as np import matplotlib.pyplot as plt def mandelbrot(c, max_iter): z = 0 n = 0 while abs(z) <= 2 and n < max_iter: z = z*z + c n += 1 return n def mandelbrot_image(xmin, xmax, ymin, ymax, width=10, height=10, max_iter=256): # Create a width x height grid of complex numbers x = np.linspace(xmin, xmax, width * 100) y = np.linspace(ymin, ymax, height * 100) X, Y = np.meshgrid(x, y) C = X + 1j * Y # Compute Mandelbrot set img = np.zeros(C.shape, dtype=int) for i in range(width * 100): for j in range(height * 100): img[j, i] = mandelbrot(C[j, i], max_iter) # Plotting plt.figure(figsize=(width, height)) plt.imshow(img, extent=(xmin, xmax, ymin, ymax), cmap='hot') plt.colorbar() plt.title(\"Mandelbrot Set\") plt.show() # Parameters defining the extent of the region in the complex plane we're going to plot xmin, xmax, ymin, ymax = -2.0, 0.5, -1.25, 1.25 width, height = 10, 10 max_iter = 256 mandelbrot_image(xmin, xmax, ymin, ymax, width, height, max_iter) Watch YouTube... from IPython.display import YouTubeVideo # Embedding the YouTube video YouTubeVideo('GtL1huin9EE', width=800, height=450) What are nibabel and nilearn? nibabel nibabel is designed to provide a simple, uniform interface to various neuroimaging file formats, allowing users to easily manipulate neuroimaging data within Python scripts or interactive environments like Jupyter notebooks. Key features and capabilities of nibabel include: Reading and Writing Neuroimaging Data: nibabel allows you to read data from disk into Python data structures and write data back to disk in various neuroimaging formats. Data Manipulation: Once loaded into Python, neuroimaging data can be manipulated just like any other data structure. This includes operations like slicing, statistical analyses, and visualization. Header Access: nibabel provides access to the headers of neuroimaging files, which contain metadata about the imaging data such as dimensions, voxel sizes, data type, and orientation. This is crucial for understanding and correctly interpreting the data. Affine Transformations: It supports affine transformations that describe the relationship between voxel coordinates and world coordinates, enabling spatial operations on the data. nilearn nilearn is a Python library designed to facilitate fast and easy statistical learning analysis and manipulation of neuroimaging data. It builds on libraries such as numpy, scipy, scikit-learn, and pandas, offering a comprehensive toolkit for neuroimaging data processing, with a focus on machine learning applications. nilearn aims to make it easier for researchers in neuroscience and machine learning to use Python for sophisticated imaging data analysis and visualization. In this intro, we won't be focusing on the more advanced uses such as those involving machine learning, but just leveraging it's ability to perform simple functions with structural MRI images. To this end, one of the strengths of nilearn is its powerful and flexible plotting capabilities, designed specifically for neuroimaging data. It provides functions to visualize MRI volumes, statistical maps, connectome diagrams, and more, with minimal code. Before we get started, we need to install the necessary packages and import the NIFTIs. We will use T1-weighted structural MRI scans, one of myself and one that I copied from OpenNeuro , a website for openly available neuroimaging datasets. # Install necessary packages %%capture !pip install nibabel matplotlib nilearn !pip install imageio # Download the MRI scan files from GitHub and rename them !wget https://raw.githubusercontent.com/sohaamir/MRICN/main/niftis/chris_T1.nii -O chris_brain.nii !wget https://raw.githubusercontent.com/sohaamir/MRICN/main/niftis/aamir_T1.nii -O aamir_brain.nii import nibabel as nib import nilearn as nil import numpy as np import pylab as plt import matplotlib.pyplot as plt import imageio import os What can we do with our data using nibabel ? Checking the data shape and type The code below loads a NIfTI image and prints out its shape and data type. The shape tells us the dimensions of the image, which typically includes the number of voxels in each spatial dimension (X, Y, Z) and sometimes time or other dimensions. The data type indicates the type of data used to store voxel values, such as float or integer types. # Load the first image using nibabel t1_aamir_path = 'aamir_brain.nii' t1_aamir_image = nib.load(t1_aamir_path) # Now let's check the image data shape and type print(\"Aamir's image shape:\", t1_aamir_image.shape) print(\"Aamir's image data type:\", t1_aamir_image.get_data_dtype()) Aamir's image shape: (192, 256, 256) Aamir's image data type: int16 Accessing the NIFTI metadata Each NIfTI file also comes with a header containing metadata about the image. Here we've extracted the voxel sizes, which represent the physical space covered by each voxel, and the image orientation, which tells us how the image data is oriented in space. # Access the image header header_aamir = t1_aamir_image.header # Print some header information print(\"Aamir's voxel size:\", header_aamir.get_zooms()) print(\"Aamir's image orientation:\", nib.aff2axcodes(t1_aamir_image.affine)) Aamir's voxel size: (0.94, 0.9375, 0.9375) Aamir's image orientation: ('R', 'A', 'S') Data visualization and comparison We can visualize the data from our NIfTI file by converting it to a numpy array and then using matplotlib to display a slice. In this example, we've displayed an axial slice from the middle of the brain, which is a common view for inspecting T1-weighted images. # Get the image data as a numpy array aamir_data = t1_aamir_image.get_fdata() # Display one axial slice of the image aamir_slice_index = aamir_data.shape[1] // 2 # Get the middle index along Z-axis plt.imshow(aamir_data[:, :, aamir_slice_index], cmap='gray') plt.title('Middle Axial Slice of Aamirs Brain') plt.axis('off') # Hide the axis to better see the image plt.show() Now we can load a second T1-weighted image and printed its shape for comparison. By comparing the shapes of the two images, we can determine if they are from the same scanning protocol or if they need to be co-registered for further analysis. # Load the second image t1_chris_path = 'chris_brain.nii' t1_chris_image = nib.load(t1_chris_path) # Let's compare the shapes of the two images print(\"First image shape:\", t1_aamir_image.shape) print(\"Second image shape:\", t1_chris_image.shape) First image shape: (192, 256, 256) Second image shape: (176, 240, 256) Now that we have loaded both T1-weighted MRI images, we are interested in visualizing and comparing them directly. To do this, we will extract the data from the second image, chris_brain.nii , and display a slice from it alongside a slice from the first image, aamir_brain.nii . # Get data for the second image chris_data = t1_chris_image.get_fdata() chris_slice_index = chris_data.shape[1] // 2 # Get the middle index along Z-axis # Display a slice from both images side by side fig, axes = plt.subplots(1, 2, figsize=(12, 6)) # Plot first image slice axes[0].imshow(aamir_data[:, :, aamir_slice_index], cmap='gray') axes[0].set_title('aamir_brain.nii') axes[0].axis('off') # Plot second image slice axes[1].imshow(chris_data[:, :, chris_slice_index], cmap='gray') axes[1].set_title('chris_brain.nii') axes[1].axis('off') plt.show() Creating Animated GIFs from MRI Scans To visualize the structure of the brain in MRI scans, we can create an animated GIF that scrolls through each slice of the scan. This is particularly useful for examining the scan in a pseudo-3D view by observing one slice at a time through the entire depth of the brain. The following code defines a function create_gif_from_mri_normalized that processes an MRI scan file and produces an animated GIF. The MRI data is first normalized by clipping the top and bottom 1% of pixel intensities, which enhances contrast and detail. The scan is then sliced along the sagittal plane, and each slice is converted to an 8-bit grayscale image and compiled into an animated GIF. This normalization process ensures that the resulting GIF maintains visual consistency across different scans. We apply this function to two MRI scans, aamir_brain.nii and chris_brain.nii , creating a GIF for each. These GIFs, named 'aamir_brain_normalized.gif' and 'chris_brain_normalized.gif', respectively, will allow us to visually assess and compare the scans. # Function to normalize and create a GIF from a 3D MRI scan in the sagittal plane def create_gif_from_mri_normalized(path, gif_name): # Load the image and get the data img = nib.load(path) data = img.get_fdata() # Normalize the data for better visualization # Clip the top and bottom 1% of pixel intensities p2, p98 = np.percentile(data, (2, 98)) data = np.clip(data, p2, p98) data = (data - np.min(data)) / (np.max(data) - np.min(data)) # Prepare to capture the slices slices = [] # Sagittal slices are along the x-axis, hence data[x, :, :] for i in range(data.shape[0]): slice = data[i, :, :] slice = np.rot90(slice) # Rotate or flip the slice if necessary slices.append((slice * 255).astype(np.uint8)) # Convert to uint8 for GIF # Create a GIF imageio.mimsave(gif_name, slices, duration=0.1) # duration controls the speed of the GIF # Create GIFs from the MRI scans create_gif_from_mri_normalized('aamir_brain.nii', 'aamir_brain_normalized.gif') create_gif_from_mri_normalized('chris_brain.nii', 'chris_brain_normalized.gif') After generating the GIFs for each MRI scan, we can now display them directly within the notebook. This visualization provides us with an interactive view of the scans, making it easier to observe the entire brain volume as a continuous animation. Below, we use the IPython.display module to render the GIFs in the notebook. The first GIF corresponds to Aamir's brain scan, and the second GIF corresponds to Chris's brain scan. These inline animations can be a powerful tool for presentations, education, and qualitative analysis, offering a dynamic view into the MRI data without the need for specialized neuroimaging software. from IPython.display import Image, display # Display the GIF for Aamir's MRI display(Image(filename='aamir_brain_normalized.gif')) # Display the GIF for Chris's MRI display(Image(filename='chris_brain_normalized.gif'))--Created by Aamir Sohail, University of Birmingham, for the Magnetic -Resonance Imaging in Cognitive Neuroscience (MRICN) module, Spring Term 2024.
-
These are the materials for my input to the MRICN module at the University of Birmingham, which consist of three main parts:
-nilearn and nibabel (not necessary at all but there if you want to learn).Here is a quick description of all three:
-![]() -
-
The aamir_vs_chris.ipynb is a Google Colab notebook demonstrating the basic functions of two Python packages, nibabel and nilearn for manipulating and plotting neuroimaging data. Nibabel is a Python library primarily used for reading and writing different types of neuroimaging data files, while Nilearn is a Python module used for conducting a wide variety of statistical analyses on neuroimaging data.
It is not necessary as part of the module to know this, and we are only scratching the surface of what nibabel and nilearn can do, but it presents an alternative approach to the conventional GUI-based alternative of FSL/FSLeyes.
Specifically, we go through the following uses:
-We then can practically demonstrate a simple case of statistical analysis by comparing the size of two brains, my own and Chris Gorgolewski's, a prime contributor to many open-source initiatives in neuroimaging, who's structural T1 scan is made publicly available through OpenNeuro.
-Ultimately, we do this both using nilearn and FSL and end up with the exact same result regardless!
Code for running the voxel comparison test in FSL:
-# Calculate the number of non-zero voxels for Chris' brain
-chris_voxels=$(fslstats chris_bet.nii -V | awk '{print $1}')
-
-# Calculate the number of non-zero voxels for Aamir's brain
-aamir_voxels=$(fslstats aamir_bet.nii -V | awk '{print $1}')
-
-# Print the number of voxels
-echo "Chris' brain size in voxels: $chris_voxels"
-echo "Aamir's brain size in voxels: $aamir_voxels"
-
-# Compare the voxel counts and print who has the bigger brain
-if [ "$chris_voxels" -gt "$aamir_voxels" ]; then
- echo "Chris has the bigger brain."
-elif [ "$chris_voxels" -lt "$aamir_voxels" ]; then
- echo "Aamir has the bigger brain."
-else
- echo "Both brains are the same size."
-fi
-![]()
This notebook interactively demonstrates the basis of functional connectivity, as we will define it in our analysis.
-Specifically, using nilearn, we can import 'resting-state' fMRI data from the Richardson dataset where 'children and adults watched Disney Pixar’s “Partly Cloudy” while lying in the scanner.
We then parcellate the regions, and take a very crude approach to functional connectivity by getting the mean time-series for each region, and then correlating that with all of the other regions in the brain. Subsequently, we can then plot the most and least-connected regions, as well as the individual time-series for the regions demonstrating the strongest and weakest correlation (or 'connectivity'):
-
The primary focus of today's workshop is to run a group-level functional connectivity analysis on resting-state fMRI data of three participants. We will specifically look at the functional connectivity of the posterior cingulate cortex (PCC), a region of the default mode network (DMN) that is commonly found to be active in resting-state data.
-To do this, we will:
-
-' + escapeHtml(summary) +'
' + noResultsText + '
'); - } -} - -function doSearch () { - var query = document.getElementById('mkdocs-search-query').value; - if (query.length > min_search_length) { - if (!window.Worker) { - displayResults(search(query)); - } else { - searchWorker.postMessage({query: query}); - } - } else { - // Clear results for short queries - displayResults([]); - } -} - -function initSearch () { - var search_input = document.getElementById('mkdocs-search-query'); - if (search_input) { - search_input.addEventListener("keyup", doSearch); - } - var term = getSearchTermFromLocation(); - if (term) { - search_input.value = term; - doSearch(); - } -} - -function onWorkerMessage (e) { - if (e.data.allowSearch) { - initSearch(); - } else if (e.data.results) { - var results = e.data.results; - displayResults(results); - } else if (e.data.config) { - min_search_length = e.data.config.min_search_length-1; - } -} - -if (!window.Worker) { - console.log('Web Worker API not supported'); - // load index in main thread - $.getScript(joinUrl(base_url, "search/worker.js")).done(function () { - console.log('Loaded worker'); - init(); - window.postMessage = function (msg) { - onWorkerMessage({data: msg}); - }; - }).fail(function (jqxhr, settings, exception) { - console.error('Could not load worker.js'); - }); -} else { - // Wrap search in a web worker - var searchWorker = new Worker(joinUrl(base_url, "search/worker.js")); - searchWorker.postMessage({init: true}); - searchWorker.onmessage = onWorkerMessage; -} diff --git a/search/search_index.json b/search/search_index.json deleted file mode 100644 index 5e7ae5a..0000000 --- a/search/search_index.json +++ /dev/null @@ -1 +0,0 @@ -{"config":{"indexing":"full","lang":["en"],"min_search_length":3,"prebuild_index":false,"separator":"[\\s\\-]+"},"docs":[{"location":"aamir_vs_chris/","text":"Who has the bigger brain, me or Chris Gorgolewski? A gentle introduction to manipulating neuroimaging data using Python with nilearn and nibabel What is Google Colab? Google Colaboratory is is a free cloud-based platform provided by Google that offers a Jupyter notebook environment for writing and executing Python code. It is primarily used for data analysis tasks but can also be used for general-purpose Python programming. It has many benefits when working with data (including neuroimaging data) such as: Free access Cloud-based hosting (available everywhere) GPU/TPU Support (lots of processing power) External Data Access (import data GitHub, Google Drive, local machine) It is an interactive environment similar to Anaconda, but with certain advantages (like those mentioned above). Similarly, Colab allows for users to run code in small chunks called 'cells', displaying any output such as images directly within the notebook as well. The programming language used by these notebooks is Python , which it organises in the form of Colab Notebooks . What can we do in Google Colab? We can do a whole bunch of things... Data visualization and plotting import matplotlib.pyplot as plt import numpy as np x = np.linspace(0, 10, 100) y = np.sin(x) plt.plot(x, y) plt.title(\"Sine Wave\") plt.xlabel(\"X\") plt.ylabel(\"sin(X)\") plt.show() import numpy as np import matplotlib.pyplot as plt def mandelbrot(c, max_iter): z = 0 n = 0 while abs(z) <= 2 and n < max_iter: z = z*z + c n += 1 return n def mandelbrot_image(xmin, xmax, ymin, ymax, width=10, height=10, max_iter=256): # Create a width x height grid of complex numbers x = np.linspace(xmin, xmax, width * 100) y = np.linspace(ymin, ymax, height * 100) X, Y = np.meshgrid(x, y) C = X + 1j * Y # Compute Mandelbrot set img = np.zeros(C.shape, dtype=int) for i in range(width * 100): for j in range(height * 100): img[j, i] = mandelbrot(C[j, i], max_iter) # Plotting plt.figure(figsize=(width, height)) plt.imshow(img, extent=(xmin, xmax, ymin, ymax), cmap='hot') plt.colorbar() plt.title(\"Mandelbrot Set\") plt.show() # Parameters defining the extent of the region in the complex plane we're going to plot xmin, xmax, ymin, ymax = -2.0, 0.5, -1.25, 1.25 width, height = 10, 10 max_iter = 256 mandelbrot_image(xmin, xmax, ymin, ymax, width, height, max_iter) Watch YouTube... from IPython.display import YouTubeVideo # Embedding the YouTube video YouTubeVideo('GtL1huin9EE', width=800, height=450) What are nibabel and nilearn? nibabel nibabel is designed to provide a simple, uniform interface to various neuroimaging file formats, allowing users to easily manipulate neuroimaging data within Python scripts or interactive environments like Jupyter notebooks. Key features and capabilities of nibabel include: Reading and Writing Neuroimaging Data: nibabel allows you to read data from disk into Python data structures and write data back to disk in various neuroimaging formats. Data Manipulation: Once loaded into Python, neuroimaging data can be manipulated just like any other data structure. This includes operations like slicing, statistical analyses, and visualization. Header Access: nibabel provides access to the headers of neuroimaging files, which contain metadata about the imaging data such as dimensions, voxel sizes, data type, and orientation. This is crucial for understanding and correctly interpreting the data. Affine Transformations: It supports affine transformations that describe the relationship between voxel coordinates and world coordinates, enabling spatial operations on the data. nilearn nilearn is a Python library designed to facilitate fast and easy statistical learning analysis and manipulation of neuroimaging data. It builds on libraries such as numpy, scipy, scikit-learn, and pandas, offering a comprehensive toolkit for neuroimaging data processing, with a focus on machine learning applications. nilearn aims to make it easier for researchers in neuroscience and machine learning to use Python for sophisticated imaging data analysis and visualization. In this intro, we won't be focusing on the more advanced uses such as those involving machine learning, but just leveraging it's ability to perform simple functions with structural MRI images. To this end, one of the strengths of nilearn is its powerful and flexible plotting capabilities, designed specifically for neuroimaging data. It provides functions to visualize MRI volumes, statistical maps, connectome diagrams, and more, with minimal code. Before we get started, we need to install the necessary packages and import the NIFTIs. We will use T1-weighted structural MRI scans, one of myself and one that I copied from OpenNeuro , a website for openly available neuroimaging datasets. # Install necessary packages %%capture !pip install nibabel matplotlib nilearn !pip install imageio # Download the MRI scan files from GitHub and rename them !wget https://raw.githubusercontent.com/sohaamir/MRICN/main/niftis/chris_T1.nii -O chris_brain.nii !wget https://raw.githubusercontent.com/sohaamir/MRICN/main/niftis/aamir_T1.nii -O aamir_brain.nii import nibabel as nib import nilearn as nil import numpy as np import pylab as plt import matplotlib.pyplot as plt import imageio import os What can we do with our data using nibabel ? Checking the data shape and type The code below loads a NIfTI image and prints out its shape and data type. The shape tells us the dimensions of the image, which typically includes the number of voxels in each spatial dimension (X, Y, Z) and sometimes time or other dimensions. The data type indicates the type of data used to store voxel values, such as float or integer types. # Load the first image using nibabel t1_aamir_path = 'aamir_brain.nii' t1_aamir_image = nib.load(t1_aamir_path) # Now let's check the image data shape and type print(\"Aamir's image shape:\", t1_aamir_image.shape) print(\"Aamir's image data type:\", t1_aamir_image.get_data_dtype()) Aamir's image shape: (192, 256, 256) Aamir's image data type: int16 Accessing the NIFTI metadata Each NIfTI file also comes with a header containing metadata about the image. Here we've extracted the voxel sizes, which represent the physical space covered by each voxel, and the image orientation, which tells us how the image data is oriented in space. # Access the image header header_aamir = t1_aamir_image.header # Print some header information print(\"Aamir's voxel size:\", header_aamir.get_zooms()) print(\"Aamir's image orientation:\", nib.aff2axcodes(t1_aamir_image.affine)) Aamir's voxel size: (0.94, 0.9375, 0.9375) Aamir's image orientation: ('R', 'A', 'S') Data visualization and comparison We can visualize the data from our NIfTI file by converting it to a numpy array and then using matplotlib to display a slice. In this example, we've displayed an axial slice from the middle of the brain, which is a common view for inspecting T1-weighted images. # Get the image data as a numpy array aamir_data = t1_aamir_image.get_fdata() # Display one axial slice of the image aamir_slice_index = aamir_data.shape[1] // 2 # Get the middle index along Z-axis plt.imshow(aamir_data[:, :, aamir_slice_index], cmap='gray') plt.title('Middle Axial Slice of Aamirs Brain') plt.axis('off') # Hide the axis to better see the image plt.show() Now we can load a second T1-weighted image and printed its shape for comparison. By comparing the shapes of the two images, we can determine if they are from the same scanning protocol or if they need to be co-registered for further analysis. # Load the second image t1_chris_path = 'chris_brain.nii' t1_chris_image = nib.load(t1_chris_path) # Let's compare the shapes of the two images print(\"First image shape:\", t1_aamir_image.shape) print(\"Second image shape:\", t1_chris_image.shape) First image shape: (192, 256, 256) Second image shape: (176, 240, 256) Now that we have loaded both T1-weighted MRI images, we are interested in visualizing and comparing them directly. To do this, we will extract the data from the second image, chris_brain.nii , and display a slice from it alongside a slice from the first image, aamir_brain.nii . # Get data for the second image chris_data = t1_chris_image.get_fdata() chris_slice_index = chris_data.shape[1] // 2 # Get the middle index along Z-axis # Display a slice from both images side by side fig, axes = plt.subplots(1, 2, figsize=(12, 6)) # Plot first image slice axes[0].imshow(aamir_data[:, :, aamir_slice_index], cmap='gray') axes[0].set_title('aamir_brain.nii') axes[0].axis('off') # Plot second image slice axes[1].imshow(chris_data[:, :, chris_slice_index], cmap='gray') axes[1].set_title('chris_brain.nii') axes[1].axis('off') plt.show() Creating Animated GIFs from MRI Scans To visualize the structure of the brain in MRI scans, we can create an animated GIF that scrolls through each slice of the scan. This is particularly useful for examining the scan in a pseudo-3D view by observing one slice at a time through the entire depth of the brain. The following code defines a function create_gif_from_mri_normalized that processes an MRI scan file and produces an animated GIF. The MRI data is first normalized by clipping the top and bottom 1% of pixel intensities, which enhances contrast and detail. The scan is then sliced along the sagittal plane, and each slice is converted to an 8-bit grayscale image and compiled into an animated GIF. This normalization process ensures that the resulting GIF maintains visual consistency across different scans. We apply this function to two MRI scans, aamir_brain.nii and chris_brain.nii , creating a GIF for each. These GIFs, named 'aamir_brain_normalized.gif' and 'chris_brain_normalized.gif', respectively, will allow us to visually assess and compare the scans. # Function to normalize and create a GIF from a 3D MRI scan in the sagittal plane def create_gif_from_mri_normalized(path, gif_name): # Load the image and get the data img = nib.load(path) data = img.get_fdata() # Normalize the data for better visualization # Clip the top and bottom 1% of pixel intensities p2, p98 = np.percentile(data, (2, 98)) data = np.clip(data, p2, p98) data = (data - np.min(data)) / (np.max(data) - np.min(data)) # Prepare to capture the slices slices = [] # Sagittal slices are along the x-axis, hence data[x, :, :] for i in range(data.shape[0]): slice = data[i, :, :] slice = np.rot90(slice) # Rotate or flip the slice if necessary slices.append((slice * 255).astype(np.uint8)) # Convert to uint8 for GIF # Create a GIF imageio.mimsave(gif_name, slices, duration=0.1) # duration controls the speed of the GIF # Create GIFs from the MRI scans create_gif_from_mri_normalized('aamir_brain.nii', 'aamir_brain_normalized.gif') create_gif_from_mri_normalized('chris_brain.nii', 'chris_brain_normalized.gif') After generating the GIFs for each MRI scan, we can now display them directly within the notebook. This visualization provides us with an interactive view of the scans, making it easier to observe the entire brain volume as a continuous animation. Below, we use the IPython.display module to render the GIFs in the notebook. The first GIF corresponds to Aamir's brain scan, and the second GIF corresponds to Chris's brain scan. These inline animations can be a powerful tool for presentations, education, and qualitative analysis, offering a dynamic view into the MRI data without the need for specialized neuroimaging software. from IPython.display import Image, display # Display the GIF for Aamir's MRI display(Image(filename='aamir_brain_normalized.gif')) # Display the GIF for Chris's MRI display(Image(filename='chris_brain_normalized.gif'))
