Snakemake pipeline for Epicure analyses: Chip-Seq, Atac-Seq, Cut&Tag, Cut&Run, MeDIP-Seq, 8-OxoG-Seq
note: This action has already been done for you if you work at Gustave Roussy. See at the end of this section
- Install snakemake and snakedeploy with mamba package manager. Given that Mamba is installed, run:
mamba create -c conda-forge -c bioconda --name bigr_epicure_pipeline snakemake snakedeploy pandas
- Ensure your conda environment is activated in your bash terminal:
conda shell.bash activate bigr_epicure_pipeline
Alternatively, if you work at Gustave Roussy, you can use our shared environment:
conda activate /mnt/beegfs/pipelines/unofficial-snakemake-wrappers/shared_install/bigr_epicure_pipeline/
note: This action has been made easier for you if you work at Gustave Roussy. See at the end of this section
Given that Snakemake and Snakedeploy are installed and available (see Installation), the workflow can be deployed as follows.
- Go to your working directory:
cd path/to/my/project
- Deploy workflow:
snakedeploy deploy-workflow https://github.com/tdayris/bigr_epicure_pipeline . --tag 0.15.0
Snakedeploy will create two folders workflow and config. The former contains the deployment of the chosen workflow as a Snakemake module, the latter contains configuration files which will be modified in the next step in order to configure the workflow to your needs. Later, when executing the workflow, Snakemake will automatically find the main Snakefile in the workflow subfolder.
- Consider to put the exact version of the pipeline and all modifications you might want perform under version control. e.g. by managing it via a (private) Github repository
Alternatively, if you work at Gustave Roussy, you can use our shared pipeline repository:
snakedeploy deploy-workflow /mnt/beegfs/pipelines/bigr_epicure_pipeline/0.15.0 . --tag 0.15.0
See dedicated config/README.md file for dedicated explanations of all options and consequences.
note: This action has been made easier for you if you work at Gustave Roussy. See at the end of this section
Given that the workflow has been properly deployed and configured, it can be executed as follows.
For running the workflow while deploying any necessary software via conda (using the Mamba package manager by default), run Snakemake with
snakemake --cores all --use-conda
Alternatively, for users at Gustave Roussy, you may use:
bash /mnt/beegfs/pipelines/bigr_epicure_pipeline/<version>/workflow/scripts/misc/bigr_launcher.sh (replace <version> accoding to your needs)
Snakemake will automatically detect the main Snakefile in the workflow subfolder and execute the workflow module that has been defined by the deployment in step 2.
For further options, e.g. for cluster and cloud execution, see the docs.
After finalizing your data analysis, you can automatically generate an interactive visual HTML report for inspection of results together with parameters and code inside of the browser via
snakemake --report report.zip
The resulting report.zip file can be passed on to collaborators, provided as a supplementary file in publications, or uploaded to a service like Zenodo in order to obtain a citable DOI.
note: The following steps may not be perform in that exact order.
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Download genome sequence | curl | Snakemake-Wrapper: download-sequence | Ensure genome sequence are consistent in Epicure analyses |
| Download genome annotation | curl | Snakemake-Wrapper: download-annotation | Ensure genome annotation are consistent in Epicure analyses |
| Download blacklised regions | wget | Ensure blacklist regions are consistent in Epicure analyses | |
| Trimming + QC | Fastp | Snakemake-Wrapper: fastp | Perform read quality check and corrections, UMI, adapter removal, QC before and after trimming |
| Quality Control | FastqScreen | Snakemake-Wrapper: fastq-screen | Perform library quality check |
| Download fastq screen indexes | wget | Ensure fastq_screen reports are the same among Epicure analyses |
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Indexation | Bowtie2 | Snakemake-Wrapper: bowtie2-build | Index genome for up-coming read mapping |
| Mapping | Bowtie2 | Snakemake-Wrapper: bowtie2-align | Align sequenced reads on the genome |
| Filtering | Sambamba | Snakemake-Wrapper: sambamba-sort | Sort alignment over chromosome position, this reduce up-coming required computing resources, and reduce alignment-file size. |
| Filtering | Sambamba | Snakemake-Wrapper: sambamba-view | Remove non-canonical chromosomes and mapping belonging to mitochondrial chromosome. Remove low quality mapping. |
| Filtering | Sambamba | Snakemake-Wrapper: sambamba-markdup | Remove sequencing duplicates. |
| Filtering | DeepTools | Snakemake-Wrapper: sambamba-sort | For Atac-seq only. Reads on the positive strand should be shifted 4 bp to the right and reads on the negative strand should be shifted 5 bp to the left as in Buenrostro et al. 2013. |
| Archive | Sambamba | Snakemake-Wrapper: sambamba-view | Compress alignment fil in CRAM format in order to reduce archive size. |
| Quality Control | Picard | Snakemake-Wrapper: picard-collect-multiple-metrics | Summarize alignments, GC bias, insert size metrics, and quality score distribution. |
| Quality Control | Samtools | Snakemake-Wrapper: samtools-stats | Summarize alignment metrics. Performed before and after mapping-post-processing in order to highlight possible bias. |
| Quality Control | DeepTools | Snakemake-Wrapper: deeptools-fingerprint | Control signal specificity. |
| GC-bias correction | DeepTools | Official Documentation | Filter regions with abnormal GC-Bias as decribed in Benjamini & Speed, 2012. OxiDIP-Seq only. |
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Coverage | DeepTools | Snakemake-Wrapper: deeptools-bamcoverage | Compute genome coverage, normalized to 1M reads |
| Coverage | MEDIPS | Official documentation | Compute genome coverage with CpG density correction using MEDIPS (MeDIP-Seq only) |
| Scaled-Coverage | DeepTools | Snakemake-Wrapper: deeptools-computematrix | Calculate scores per genomic regions. Used for heatmaps and profile coverage plots. |
| Depth | DeepTools | Snakemake-Wrapper: deeptools-plotheatmap | Assess the sequencing depth of given samples |
| Coverage | CSAW | Official documentation | Count and filter reads over sliding windows. |
| Filter | CSAW | Official documentation | Filter reads over background signal. |
| Quality Control | DeepTools | Snakemake-Wrapper: deeptools-plotprofile | Plot profile scores over genomic regions |
| Quality Control | DeepTools | Official Documentation | Plot principal component analysis (PCA) over bigwig coverage to assess sample dissimilarity. |
| Quality Control | DeepTools | Official Documentation | Plot sample correlation based on the coverage analysis. |
| Coverage | BedTools | Snakemake-Wrapper: bedtools-genomecov | Estimate raw coverage over the genome |
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Peak-Calling | Mac2 | Snakemake-Wrapper: macs2-callpeak | Search for significant peaks |
| Heatmap | DeepTools | Snakemake-Wrapper: deeptools-plotheatmap | Plot heatmap and peak coverage among samples |
| FRiP score | Manually assessed | Compute Fragment of Reads in Peaks to assess signal/noise ratio. | |
| Peak-Calling | SEACR | Official Documentation | Search for significant peaks in Cut&Tag or Cut&Run |
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Peak-Calling | MEDIPS | Official documentation | Search for significant variation in peak coverage with EdgeR (MeDIP-Seq only) |
| Normalization | CSAW | Official documentation | Correct composition bias and variable library sizes. |
| Differential Binding | CSAW-EdgeR | Official documentation | Call for differentially covered windows |
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Annotation | MEDIPS | Official Documentation | Annotate peaks with Ensembl-mart through MEDIPS (MeDIP-Seq only) |
| Annotation | Homer | Snakemake-Wrapper: homer-annotatepeaks | Performing peak annotation to associate peaks with nearby genes. |
| Annotation | CHiP seeker | Official Documentation | Performing peak annotation to associate peaks with nearby genes. |
| Quality Control | CHiP Seeker | Official Documentation | Perform region-wise barplot graph. |
| Quality Control | CHiP Seeker | Official Documentation | Perform region-wise upset-plot. |
| Quality Control | CHiP Seeker | Official Documentation | Visualize distribution of TF-binding loci relative to TSS |
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| De-novo | Homer | Official Documentation | Perform de novo motifs discovery. |

Reads are trimmed using fastp version 0.23.2, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using bowtie2 version 2.5.1, using --very-sensitive parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using Sambamba version 1.0 with parameter --filter 'mapping_quality >= 30'. In case of pair-ended library, orphan reads a dropped out using --filter 'not (unmapped or mate_is_unmapped)' in Sambamba version 1.0. Remaining reads are filtered over canonical chromosomes, using Samtools version 1.17. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome, BedTools version 2.31.0 with -wa -sorted as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using Sambamba version 1.0 using the optional parameter --remove-duplicates --overflow-list-size 600000.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0, and Samtools version 1.17.
Genome coverage was assessed with DeepTools version 3.5.2. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters --normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by Macs2 version 2.2.9.1 using both broad and narrow options. Pair-ended libraries recieved the --format BAMPE parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0, using Org.eg.db annotations version 3.16.0.
De novo motif discovery was made with Homer version 4.11 over called peaks, using the following optional parameters -size 200 -homer2 over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using CSAW version 1.32.0. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in CSAW documentation, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The prior N was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3. Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.6.0.
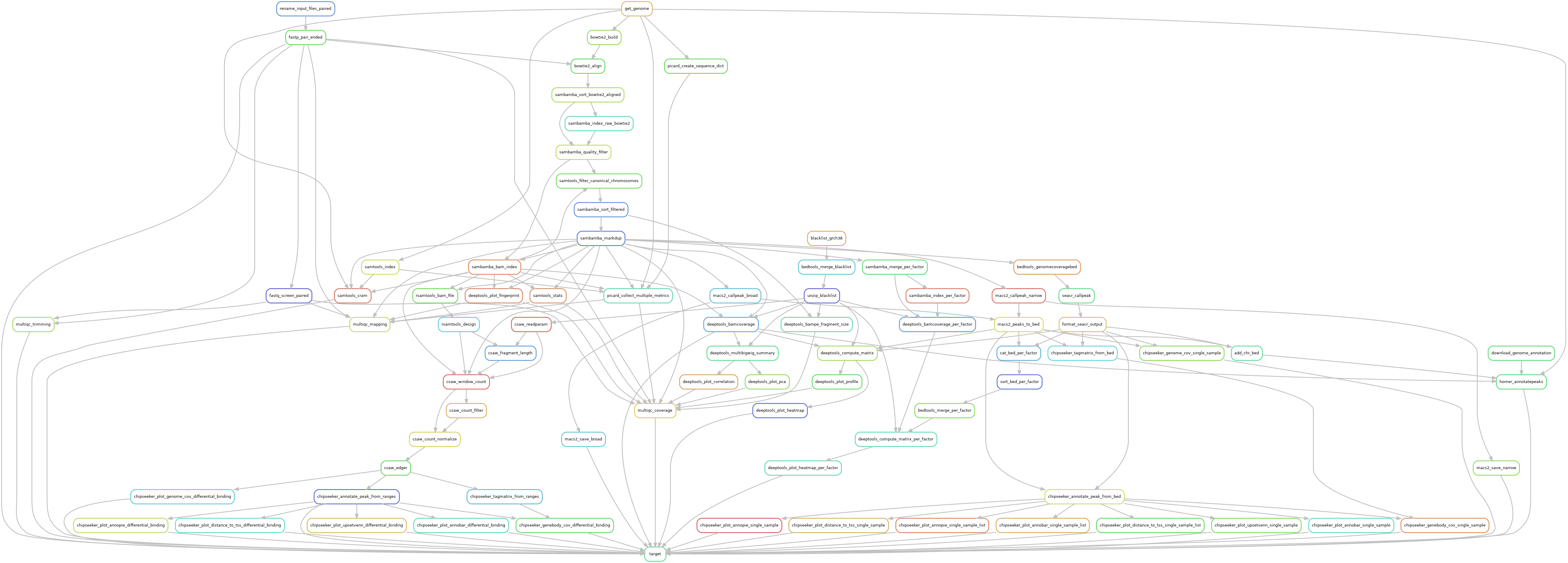
Reads are trimmed using fastp version 0.23.2, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using bowtie2 version 2.5.1, using --very-sensitive parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using Sambamba version 1.0 with parameter --filter 'mapping_quality >= 30'. In case of pair-ended library, orphan reads a dropped out using --filter 'not (unmapped or mate_is_unmapped)' in Sambamba version 1.0. Remaining reads are filtered over canonical chromosomes, using Samtools version 1.17. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome, BedTools version 2.31.0 with -wa -sorted as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using Sambamba version 1.0 using the optional parameter --remove-duplicates --overflow-list-size 600000.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0, and Samtools version 1.17.
Genome coverage was assessed with DeepTools version 3.5.2. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. MNase specificity was also taken into account using the following optional parameters --normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY --MNase. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by Macs2 version 2.2.9.1 using both broad and narrow options. Pair-ended libraries recieved the --format BAMPE parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0, using Org.eg.db annotations version 3.16.0.
De novo motif discovery was made with Homer version 4.11 over called peaks, using the following optional parameters -size 200 -homer2 over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using CSAW version 1.32.0. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in CSAW documentation, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The prior N was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3. Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.6.0.
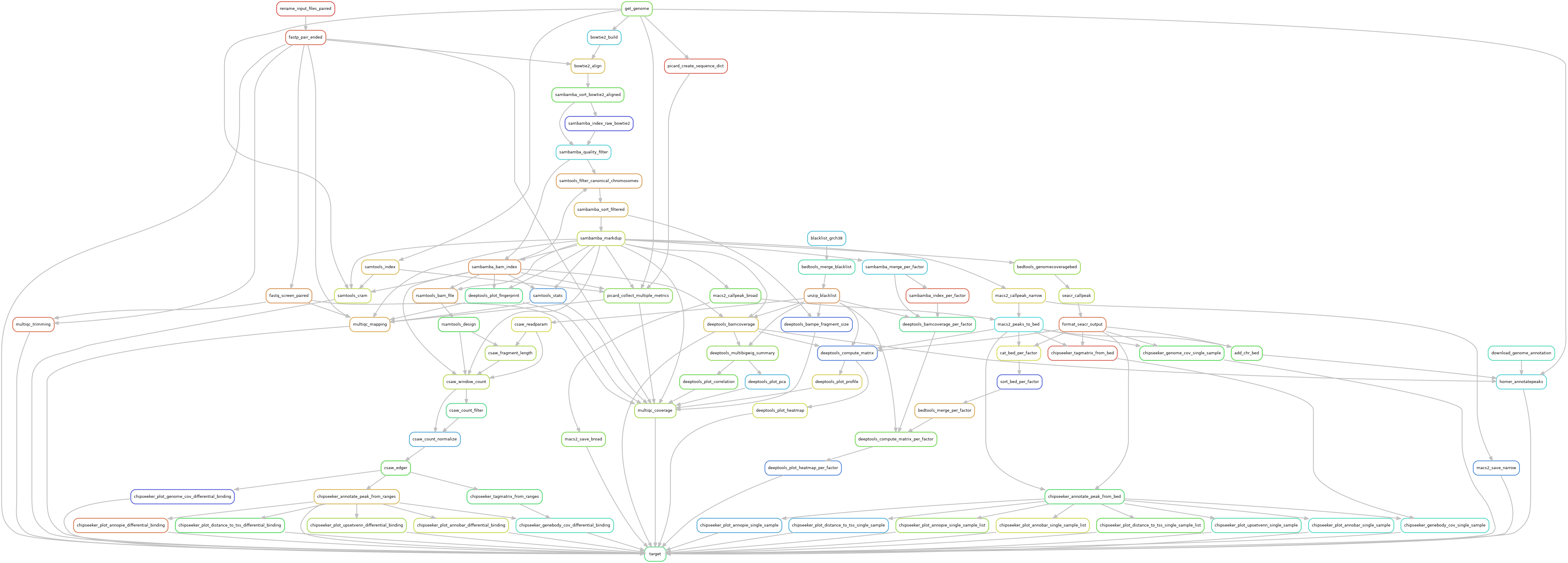
Reads are trimmed using fastp version 0.23.2, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using bowtie2 version 2.5.1, using --very-sensitive parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using Sambamba version 1.0 with parameter --filter 'mapping_quality >= 30'. In case of pair-ended library, orphan reads a dropped out using --filter 'not (unmapped or mate_is_unmapped)' in Sambamba version 1.0. Remaining reads are filtered over canonical chromosomes, using Samtools version 1.17. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome, BedTools version 2.31.0 with -wa -sorted as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using Sambamba version 1.0 using the optional parameter --remove-duplicates --overflow-list-size 600000.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0, and Samtools version 1.17.
Genome coverage was assessed with DeepTools version 3.5.2. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. Gro-seq specificity was also taken into account using the following optional parameters --normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY --Offset 12. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by Macs2 version 2.2.9.1 using both broad and narrow options. Pair-ended libraries recieved the --format BAMPE parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0, using Org.eg.db annotations version 3.16.0.
De novo motif discovery was made with Homer version 4.11 over called peaks, using the following optional parameters -size 200 -homer2 over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using CSAW version 1.32.0. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in CSAW documentation, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The prior N was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3. Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.6.0.
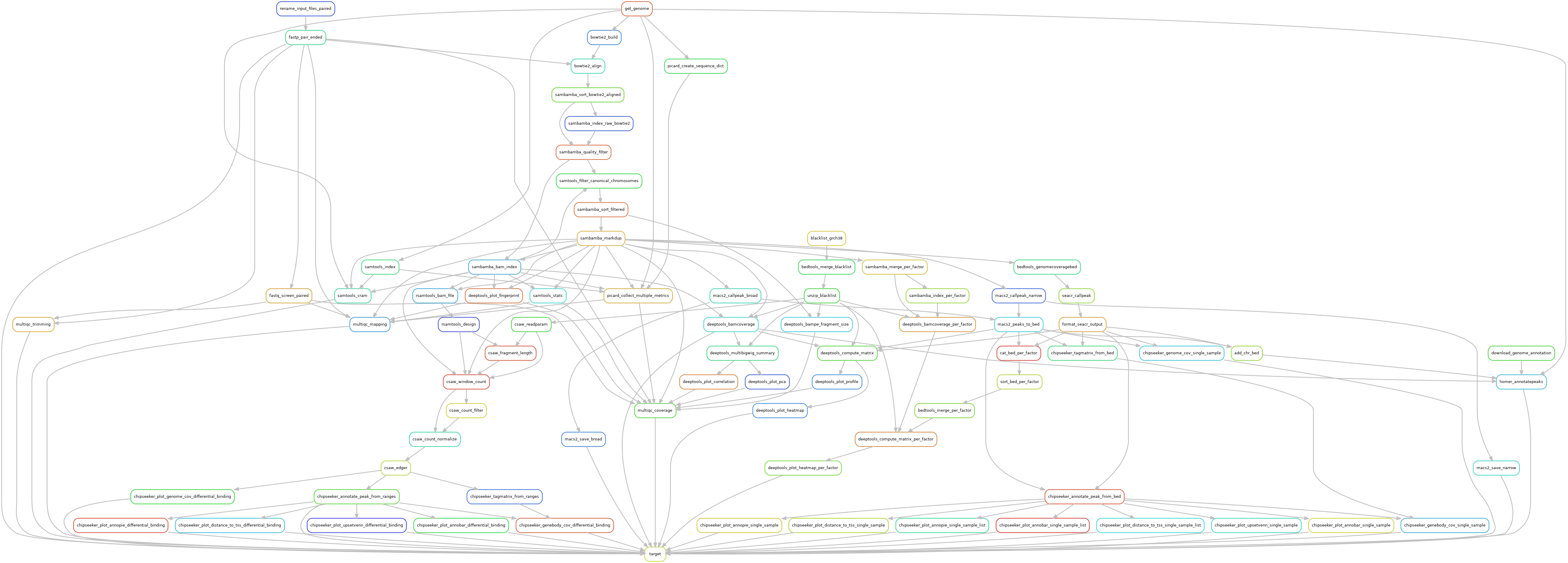
Reads are trimmed using fastp version 0.23.2, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using bowtie2 version 2.5.1, using --very-sensitive parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using Sambamba version 1.0 with parameter --filter 'mapping_quality >= 30'. In case of pair-ended library, orphan reads a dropped out using --filter 'not (unmapped or mate_is_unmapped)' in Sambamba version 1.0. Remaining reads are filtered over canonical chromosomes, using Samtools version 1.17. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome, BedTools version 2.31.0 with -wa -sorted as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using Sambamba version 1.0 using the optional parameter --remove-duplicates --overflow-list-size 600000.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0, and Samtools version 1.17.
Genome coverage was assessed with DeepTools version 3.5.2. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. Ribo-seq specificity was also taken into account using the following optional parameters --normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY --Offset 15. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by Macs2 version 2.2.9.1 using both broad and narrow options. Pair-ended libraries recieved the --format BAMPE parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0, using Org.eg.db annotations version 3.16.0.
De novo motif discovery was made with Homer version 4.11 over called peaks, using the following optional parameters -size 200 -homer2 over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using CSAW version 1.32.0. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in CSAW documentation, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The prior N was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3. Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.6.0.

Reads are trimmed using fastp version 0.23.2, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using bowtie2 version 2.5.1, using --very-sensitive parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using Sambamba version 1.0 with parameter --filter 'mapping_quality >= 30'. In case of pair-ended library, orphan reads a dropped out using --filter 'not (unmapped or mate_is_unmapped)' in Sambamba version 1.0. Remaining reads are filtered over canonical chromosomes, using Samtools version 1.17. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome, BedTools version 2.31.0 with -wa -sorted as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using Sambamba version 1.0 using the optional parameter --remove-duplicates --overflow-list-size 600000. Remaining reads were shifted as described in Buenrostro et al. 2013 : reads on the positive strand were shifted 4 bp to the right and reads on the negative were be shifted 5 bp to the left using DeepTools version 3.5.2 and the optional parameter --ATACShift.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0, and Samtools version 1.17.
Genome coverage was assessed with DeepTools version 3.5.2. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters --normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by Macs2 version 2.2.9.1 using both broad and narrow options. Pair-ended libraries recieved the --format BAMPE parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0, using Org.eg.db annotations version 3.16.0.
De novo motif discovery was made with Homer version 4.11 over called peaks, using the following optional parameters -size 200 -homer2 over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using CSAW version 1.32.0. A large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in CSAW documentation, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The prior N was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3. Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.6.0.

Reads are trimmed using fastp version 0.23.2, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using bowtie2 version 2.5.1, using --very-sensitive parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using Sambamba version 1.0 with parameter --filter 'mapping_quality >= 30'. In case of pair-ended library, orphan reads a dropped out using --filter 'not (unmapped or mate_is_unmapped)' in Sambamba version 1.0. Remaining reads are filtered over canonical chromosomes, using Samtools version 1.17. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome, BedTools version 2.31.0 with -wa -sorted as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were not filtered out, but marked as duplicates, using Sambamba version 1.0 using the optional parameter --overflow-list-size 600000. Remaining reads were shifted as described in Buenrostro et al. 2013 : reads on the positive strand were shifted 4 bp to the right and reads on the negative were be shifted 5 bp to the left using DeepTools version 3.5.2 and the optional parameter --ATACShift.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0, and Samtools version 1.17.
Genome coverage was assessed with DeepTools version 3.5.2. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters --normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY --ignoreDuplicates. Reads marked as duplicates were treated as normal reads. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by Macs2 version 2.2.9.1 using both broad and narrow options. Pair-ended libraries recieved the --format BAMPE parameters, while single-ended libraries used estimated fragment size provided in input. Alongside with Macs2, SEACR version 1.3 was used to perform single-sample peak-calling using parameters hinted in the original publication of SEACR.
The peak annotation was done using ChIPSeeker version 1.34.0, using Org.eg.db annotations version 3.16.0.
De novo motif discovery was made with Homer version 4.11 over called peaks, using the following optional parameters -size 200 -homer2 over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using CSAW version 1.32.0. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in CSAW documentation, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The prior N was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Through the whole process, reads flagged as duplicates are treated as any other read. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3. Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.6.0.

Reads are trimmed using fastp version 0.23.2, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using bowtie2 version 2.5.1, using --very-sensitive parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using Sambamba version 1.0 with parameter --filter 'mapping_quality >= 30'. In case of pair-ended library, orphan reads a dropped out using --filter 'not (unmapped or mate_is_unmapped)' in Sambamba version 1.0. Remaining reads are filtered over canonical chromosomes, using Samtools version 1.17. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome, BedTools version 2.31.0 with -wa -sorted as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were not filtered out, but marked as duplicates, using Sambamba version 1.0 using the optional parameter --overflow-list-size 600000.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0, and Samtools version 1.17.
Genome coverage was assessed with DeepTools version 3.5.2. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters --normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY --ignoreDuplicates. Reads marked as duplicates were treated as normal reads. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by Macs2 version 2.2.9.1 using both broad and narrow options. Pair-ended libraries recieved the --format BAMPE parameters, while single-ended libraries used estimated fragment size provided in input.Alongside with Macs2, SEACR version 1.3 was used to perform single-sample peak-calling using parameters hinted in the original publication of SEACR.
The peak annotation was done using ChIPSeeker version 1.34.0, using Org.eg.db annotations version 3.16.0.
De novo motif discovery was made with Homer version 4.11 over called peaks, using the following optional parameters -size 200 -homer2 over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using CSAW version 1.32.0. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in CSAW documentation, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The prior N was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Through the whole process, reads flagged as duplicates are treated as any other read. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3. Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.6.0.

Reads are trimmed using fastp version 0.23.2, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using bowtie2 version 2.5.1, using --very-sensitive parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using Sambamba version 1.0 with parameter --filter 'mapping_quality >= 30'. In case of pair-ended library, orphan reads a dropped out using --filter 'not (unmapped or mate_is_unmapped)' in Sambamba version 1.0. Remaining reads are filtered over canonical chromosomes, using Samtools version 1.17. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome, BedTools version 2.31.0 with -wa -sorted as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using Sambamba version 1.0 using the optional parameter --remove-duplicates --overflow-list-size 600000.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0, and Samtools version 1.17.
Genome coverage was assessed with DeepTools version 3.5.2. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters --normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by Macs2 version 2.2.9.1 using both broad and narrow options. Pair-ended libraries recieved the --format BAMPE parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0, using Org.eg.db annotations version 3.16.0.
De novo motif discovery was made with Homer version 4.11 over called peaks, using the following optional parameters -size 200 -homer2 over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed with MEDIPS version 1.50.0. If available, input signals were used to search signal of interest over the background noise. An adjusted p-value threshold of 0.1 was chosen to find significant signal over noise. A distance of 1 base was used to merge neighboring significant windows. Differentially binded sites were called using EdgeR and FDR were corrected using MEDIPS internal methods as defined in the official documentation. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3. Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.6.0.

Reads are trimmed using fastp version 0.23.2, using the following non-default parameter: --poly_g_min_len 25 since we expect a higher number of base modifications. Trimmed reads are mapped over the genome of interest defined in the configuration file, using bowtie2 version 2.5.1, using default parameters.
Mapped reads with a quality lower than 30 are dropped out using Sambamba version 1.0 with parameter --filter 'mapping_quality >= 30'. In case of pair-ended library, orphan reads a dropped out using --filter 'not (unmapped or mate_is_unmapped)' in Sambamba version 1.0. Remaining reads are filtered over canonical chromosomes, using Samtools version 1.17. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome, BedTools version 2.31.0 with -wa -sorted as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using Sambamba version 1.0 using the optional parameter --remove-duplicates --overflow-list-size 600000. GC bias was estimated and corrected using DeepTools version 3.5.2 using default parameters.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0, and Samtools version 1.17.
Genome coverage was assessed with DeepTools version 3.5.2. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters --normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by Macs2 version 2.2.9.1 using both broad and narrow options. Pair-ended libraries recieved the --format BAMPE parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0, using Org.eg.db annotations version 3.16.0.
De novo motif discovery was made with Homer version 4.11 over called peaks, using the following optional parameters -size 200 -homer2 over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using CSAW version 1.32.0. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in CSAW documentation, since libraries of the same size can still have composition bias, and 8oxoG changes the base composition. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The prior N was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Through the whole process, reads flagged as duplicates are treated as any other read. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3. Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.6.0.
- Fastp:
Chen, Shifu, et al. "fastp: an ultra-fast all-in-one FASTQ preprocessor." Bioinformatics 34.17 (2018): i884-i890. - Bowtie2:
Langmead, Ben, and Steven L. Salzberg. "Fast gapped-read alignment with Bowtie 2." Nature methods 9.4 (2012): 357-359. - Sambamba:
Tarasov, Artem, et al. "Sambamba: fast processing of NGS alignment formats." Bioinformatics 31.12 (2015): 2032-2034. - Ensembl:
Martin, Fergal J., et al. "Ensembl 2023." Nucleic Acids Research 51.D1 (2023): D933-D941. - Snakemake:
Köster, Johannes, and Sven Rahmann. "Snakemake—a scalable bioinformatics workflow engine." Bioinformatics 28.19 (2012): 2520-2522. - Atac-Seq:
Buenrostro, Jason D., et al. "Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position." Nature methods 10.12 (2013): 1213-1218. - MeDIPS:
Lienhard, Matthias, et al. "MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments." Bioinformatics 30.2 (2014): 284-286. - DeepTools:
Ramírez, Fidel, et al. "deepTools: a flexible platform for exploring deep-sequencing data." Nucleic acids research 42.W1 (2014): W187-W191. - UCSC:
Nassar LR, Barber GP, Benet-Pagès A, Casper J, Clawson H, Diekhans M, Fischer C, Gonzalez JN, Hinrichs AS, Lee BT, Lee CM, Muthuraman P, Nguy B, Pereira T, Nejad P, Perez G, Raney BJ, Schmelter D, Speir ML, Wick BD, Zweig AS, Haussler D, Kuhn RM, Haeussler M, Kent WJ. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 2022 Nov 24;. PMID: 36420891 - ChIPSeeker:
Yu, Guangchuang, Li-Gen Wang, and Qing-Yu He. "ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization." Bioinformatics 31.14 (2015): 2382-2383. - CSAW:
Lun, Aaron TL, and Gordon K. Smyth. "csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows." Nucleic acids research 44.5 (2016): e45-e45. - Macs2:
Gaspar, John M. "Improved peak-calling with MACS2." BioRxiv (2018): 496521. - Homer:
Heinz S, Benner C, Spann N, Bertolino E et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell 2010 May 28;38(4):576-589. PMID: 20513432 - Seacr:
Meers, Michael P., Dan Tenenbaum, and Steven Henikoff. "Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling."Epigenetics & chromatin 12 (2019): 1-11. - BedTools:
Quinlan, Aaron R., and Ira M. Hall. "BEDTools: a flexible suite of utilities for comparing genomic features." Bioinformatics 26.6 (2010): 841-842. - Snakemake-Wrappers version v2.6.0
- Snakemake-Workflows
- BiGR Epicure
- Coverage: PBC
- Peak-annotation: CentriMo
- Differential Peak Calling: DiffBind
- IGV: screen-shot, igv-reports
Based on Snakemake-Wrappers version v2.6.0