Releases: ktmeaton/ncov-recombinant
v0.7.0 - Recursive Recombinants
Notes
This is a minor release aimed towards a nextclade dataset upgrade from 2022-10-27 to 2023-01-09 which adds nomenclature for newly designated recombinants XBH - XBP. This release also adds initial support for the detection of "recursive recombination" including XBL and XBN which are recombinants of XBB.
A comprehensive test summary report can be downloaded directly with: ncov-recombinant_v0.6.1_v0.7.0.zip or viewed at the following link once the release is complete.
Documentation
- Issue #24: Create documentation on Read The Docs
Dataset
- Issue #210: Handle numeric strain names.
Resources
- Issue #185: Simplify creation of the pango-lineage nomenclature phylogeny to use the lineage_notes.txt file and the pango_aliasor library.
sc2rf
- Issue #195: Add bypass to intermission allele ratio for edge cases.
- Issue #204: Add special handling for XBB sequenced with ARTIC v4.1 and dropout regions.
- Issue #205: Add new column
parents_conflictto indicate whether the reported lineages from covSPECTRUM conflict with the reported parental clades from `sc2rf. - Issue #213: Add
XBKto auto-pass lineages. - Issue #222: Add new parameter
--gisaid-access-keytosc2rfandsc2rf_recombinants. - Issue #229: Fix bug where auto-pass lineages are missing when exclude_negatives is set to true.
- Issue #231: Fix bug where 'null' lineages in covSPECTRUM caused error in
sc2rfpostprocess. - The order of the
postprocessing.pywas rearranged to have more comprehensive details for auto-pass lineages. - Add
XANto auto-pass lineages.
Plot
- Issue #209: Restrict the palette for
rbd_levelto the range of0:12. - Issue #218: Fix bug concerning data fragmentation with large numbers of sequences.
- Issue #221: Remove parameter
--singletonsin favor of--min-cluster-sizeto control cluster size in plots. - Issue #224: Fix bug where plot crashed with extremely large datasets.
- Combine
plotandplot_historicalinto one snakemake rule. Also at custom patternplot_NX(ex.plot_N10) to adjust min cluster size.
Report
- Combine
reportandreport_historicalinto one snakemake rule.
Validate
- Issue #225: Fix bug where false negatives passed validation because the status column wasn't checked.
Designated Lineages
- Issue #217:
XBB.1.5 - Issue #196:
XBF - Issue #206:
XBG - Issue #196:
XBH - Issue #199:
XBJ - Issue #213:
XBK - Issue #219:
XBL - Issue #215:
XBM - Issue #197:
XBN
Proposed Lineages
- Issue #203:
proposed1305 - Issue #208:
proposed1340 - Issue #212:
proposed1425 - Issue #214:
proposed1440 - Issue #216:
proposed1444 - Issue #220:
proposed1576
Commits
c279f1e4docs: add changelog for v0.7.02964b4a1docs: update notes to include 1576 proposed issuefdc874abdocs: add test summary package for v0.7.03f3d4438docs: update docs v0.7.078696b36script: add bug fix to sc2rf postprocess for #231403777a0script: lint plotting script2a09c783script: fix sc2rf postprocess bug in duplicate removald44d5f90data: add XBP to controls-gisaid4293439cprofile: add controls-gisaid to virusseq builds91d6fb89defaults: update nextclade dataset to 2023-02-01630b2cd5resources: update49e6f598profile: add virusseq profile7e586d1dscript: add extra logic for auto-passing lineages0ebe5e9cscript: fix bug in report where it didn't check that plots existed25b2f243docs: update developers guide914d933fdefaults: add XBN to controls-gisaid and validation8eaf08a9data: restore controls-gisaid strain listfa123009script: defragment plot for 2185f24f695dataset: update controls-gisaid strain listefc5aab7defaults: update validation to fix XBH dropout- See CHANGELOG.md for additional commits.
v0.6.1 - Network Stability and False Positives
v0.6.1
Notes
This is a minor bugfix release aimed towards resolving network connectivity errors and catching false positives.
sc2rf
- Issue #195: Consider alleles outside of parental regions as intermissions (conflicts) to catch false positives.
- Issue #201: Make LAPIS query of covSPECTRUM optional, to help with users with network connectivity issues. This can be set with the flag
lapis: falsein builds under the rulesc2rf_recombinants. - Issue #202: Document connection errors related to LAPIS and provide options for solutions.
Commits
83ee0139docs: update changelog for v0.6.100fe2fc8docs: update notes for v0.6.1fa03ea96workflow: fix bug where rbd_levels log was incorrectly nameda281b75cworkflow: make lapis optional param for #201 #20275684b55docs: update docs1085ce0escript: postprocess count alleles outside regions as intermissions for #195c11770c1param: add XAV to auto-pass for #104 #195
v0.6.0 - Sublineages and Immunity
v0.6.0
Notes
This is a major release that includes the following changes:
-
Detection of all recombinants in Nextclade dataset 2022-10-27:
XAtoXBE. -
Implementation of recombinant sublineages (ex.
XBB.1). -
Implementation of immune-related statistics (
rbd_level,immune_escape,ace2_binding) fromnextclade, theNextstrainteam, and Jesse Bloom's group:- https://github.com/nextstrain/ncov/blob/master/defaults/rbd_levels.yaml
- https://jbloomlab.github.io/SARS-CoV-2-RBD_DMS_Omicron/epistatic-shifts/
- https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/escape-calc/
- https://doi.org/10.1093/ve/veac021
- https://doi.org/10.1101/2022.09.15.507787
- https://doi.org/10.1101/2022.09.20.508745
Dataset
- Issue #168: NULL collection dates and NULL country is implemented.
controlswas updated to in include 1 strain fromXBBfor a total of 22 positive controls. The 28 negative controls were unchanged fromv0.5.1.controls-gisaidstrain list was updated to includeXAthrough toXBEfor a total of 528 positive controls. This includes sublineages such asXBB.1andXBB.1.2which synchronizes with Nextclade Dataset 2022-10-19. The 187 negatives controls were unchanged fromv0.5.1.
Nextclade
- Issue #176: Upgrade Nextclade dataset to tag
2022-10-27and upgrade Nextclade tov2.8.0. - Issue #193: Use the nextclade dataset
sars-cov-2-21Lto calculateimmune_escapeandace2_binding.
RBD Levels
- Issue #193: Create new rule
rbd_levelsto calculate the number of key receptor binding domain (RBD) mutations.
Lineage Tree
- Issue #185: Use nextclade dataset Auspice tree for lineage hierarchy. Previously, the phylogeny of lineages was constructed from the cov-lineages website YAML. Instead, we now use the tree provided with nextclade datasets, to better synchronize the lineage model with the output.
Rather than creating the output tree in resources/lineages.nwk, the lineage tree will output to data/sars-cov-2_<DATE>/tree.nwk. This is because different builds might use different nextclade datasets, and so are dataset specific output.
sc2rf
- Issue #179: Fix bug where
sc2rf/recombinants.ansi.txtis truncated. - Issue #180: Fix recombinant sublineages (ex. XAY.1) missing their derived mutations in the
cov-spectrum_query. Previously, thecov-spectrum_querymutations were only based on the parental alleles (before recombination). This led to sublinaeges (ex.XAY.1,XAY.2) all having the exact same query. Now, thecov-spectrum_querywill include all substitutions shared between all sequences in thecluster_id. - Issue #187: Document bug that occurs if duplicate sequences are present, and the initial validation was skipped by not running
scripts/create_profile.sh. - Issue #191 and Issue #192: Reduce false positives by ensuring that each mode of sc2rf has at least one additional parental population that serves as the alternative hypothesis.
- Issue #195: Implement a filter on the ratio of intermissions to alleles. Sequences will be marked as false positives if the number of intermissions (i.e. alleles that conflict with the identified parental region) is greater than or equal to the number of alleles contributed by the minor parent. This ratio indicates that there is more evidence that conflicts with recombination than there is allele evidence that supports a recombinant origin.
Linelist
- Issue #183: Recombinant sublineages. When nextclade calls a lineage (ex.
XAY.1) which is a sublineage of a sc2rf lineage (XAY), we prioritize the nextclade assignment. - Issue #193: Add immune-related statistics:
rbd_levels,rbd_substitutions,immune_escape, andace2_binding.
Plot
- Issue #57: Include substitutions within breakpoint intervals for breakpoint plots. This is a product of Issue #180 which provides access to all substitutions.
- Issue #112: Fix bug where breakpoints plot image was out of bounds.
- Issue #188: Remove the breakpoints distribution axis (ex.
breakpoints_clade.png) in favor of putting the legend at the top. This significant reduces plotting issues (ex. Issue #112). - Issue #193: Create new plot
rbd_level.
Validate
Designated Lineages
- Issue #85:
XAY, updated controls - Issue #178:
XAY.1 - Issue #172:
XBB.1 - Issue #175:
XBB.1.1 - Issue #184:
XBB.1.2 - Issue #173:
XBB.2 - Issue #174:
XBB.3 - Issue #181:
XBC.1 - Issue #182:
XBC.2 - Issue #171:
XBD - Issue #177:
XBE
Proposed Lineages
- Issue #198:
proposed1229 - Issue #199:
proposed1268 - Issue #197:
proposed1296
Commits
2506e907docs: update changelog and add v0.6.0 testing summary package0cc421e0docs: update all contributorscd9b6cbbresources: update issues0fa2e3c1docs: update readme375c3a76resources: add proposed lineages for #197 #198 #199dad989e7param: remove BQ.1 from sc2rf mode VOC as its too close to BA.5.3d7cb005fdocs: update issue template lineage-validation1beac97eresources: add XBF to curated breakpoints for #196fae7bfdbscript: sc2rf implement intermission allele ratio for #19589a41265script: additional manual curation of lineage_treeebd3ce1fresources: update validation strains for controls-gisaidd8bff572script: add RBD Level slide to reportc1879c1dscript: catch errors in rbd_level plotting with no recombinants63545a08script: fix bug in linelist with cluster_privatesc24a7179resources: update issuesd32d557fdocs: update development notes7f825a41script: manual fix for CK in lineage_treefdd6f66dworkflow: implement rbd levels for #1930058dd6eparam: upgrade nextclade dataset to 2022-10-27 and reduce breakpoints of XA modefb062c32env: upgrade nextclade to v2.8.0- See CHANGELOG.md for additional commits.
v0.5.1 - Hotfix
v0.5.1
This hotfix release fixes Issue #169 which was caused by an internal change in snakemake regarding dependencies. This was resolved by version controlling the tabulate package.
Notes
Workflow
- Issue #169: AttributeError: 'str' object has no attribute 'name'
Resources
- Issue #167: Alias key out of date, change source
Validate
Proposed Lineages
- Issue #166:
proposed1138 - Issue #165:
proposed1139
Commits
v0.5.0 - XA to XBC
v0.5.0
Please check out the
v0.5.0Testing Summary Package for a comprehensive report.
Notes
This is a minor release that includes the following changes:
- Detection of all recombinants in Nextclade dataset 2022-09-27:
XAtoXBC. - Create any number of custom
sc2rfmodes with CLI arguments.
Resources
- Issue #96: Create newick phylogeny of pango lineage parent child relationships, to get accurate sublineages including aliases.
- Issue #118: Fix missing pango-designation issues for XAY and XBA.
Datasets
- Issue #25: Reduce positive controls to one sequence per clade. Add new positive controls
XAL,XAP,XAS,XAU, andXAZ. - Issue #92: Reduce negative controls to one sequence per clade. Add negative control for
22D (Omicron) / BA.2.75. - Issue #155: Add new profile and dataset
controls-gisaid. Only a list of strains is provided, as GISAID policy prohibits public sharing of sequences and metadata.
Profile Creation
- Issue #77: Report slurm command for
--hpcprofiles inscripts/create_profiles.sh. - Issue #153: Fix bug where build parameters
metadataandsequenceswere not implemented.
Nextclade
sc2rf
-
Issue #78: Add new parameter
max_breakpoint_lentosc2rf_recombinantsto mark samples with two much uncertainty in the breakpoint interval as false positives. -
Issue #79: Add new parameter
min_consec_alleletosc2rf_recombinantsto ignore recombinant regions with less than this number of consecutive alleles (both diagnostic SNPs and diganostic reference alleles). -
Issue #80: Migrate sc2rf froma submodule to a subdirectory (including LICENSE!). This is to simplify the updating process and avoid errors where submodules became out of sync with the main pipeline.
-
Issue #83: Improve error handling in
sc2rf_recombinantswhen the input stats files are empty. -
Issue #89: Reduce the default value of the parameter
min_leninsc2rf_recombinantsfrom1000to500.This is to handleXAPandXAJ. -
Issue #90: Auto-pass select nextclade lineages through
sc2rf:XN,XP,XAR,XAS, andXAZ. This requires differentiating the nextclade inputs as separate parameters--nextcladeand--nextclade-no-recom.-
XN,XP, andXARhave extremely small recombinant regions at the terminal ends of the genome. Depending on sequencing coverage,sc2rfmay not reliably detect these lineages. -
The newly designated
XASandXAZpose a challenge for recombinant detection using diagnostic alleles. The first region ofXAScould be eitherBA.5orBA.4based on subsitutions, but is mostly likelyBA.5based on deletions. Since the region contains no diagnostic alleles to discriminateBA.5vs.BA.4, breakpoints cannot be detected bysc2rf. -
Similarly for
XAZ, theBA.2segments do not contain anyBA.2diagnostic alleles, but instead are all reversion fromBA.5alleles. TheBA.2parent was discovered by deep, manual investigation in the corresponding pango-designation issue. Since theBA.2regions contain no diagnostic forBA.2, breakpoints cannot be detected bysc2rf.
-
-
Issue #95: Generalize
sc2rf_recombinantsto take any number of ansi and csv input files. This allows greater flexibility in command-line arguments tosc2rfand are not locked into the hardcodedprimaryandsecondaryparameter sets. -
Issue #96: Include sub-lineage proportions in the
parents_lineage_confidence. This reduces underestimating the confidence of a parental lineage. -
Issue #150: Fix bug where
sc2rfwould write empty output csvfiles if no recombinants were found. -
Issue #151: Fix bug where samples that failed to align were missing from the linelists.
-
Issue #158: Reduce
sc2rfparam--max-intermission-lengthfrom3to2to be consistent with Issue #79. -
Issue #161: Implement selection method to pick best results from various
sc2rfmodes. -
Issue #162: Upgrade
sc2rf/virus_properties.json. -
Issue #163: Use LAPIS
nextcladePangoLineageinstead ofpangoLineage. Also disable default filtermax_breakpoint_lenforXAN. -
Issue #164: Fix bug where false positives would appear in the filter
sc2rfansi output (recombinants.ansi.txt). -
The optional
lapisparameter forsc2rf_recombinantshas been removed. Querying LAPIS for parental lineages is no longer experimental and is now an essential component (cannot be disabled). -
The mandatory
mutation_thresholdparameter forsc2rfhas been removed. Instead,--mutation-thresholdcan be set independently in each of thescrfmodes.
Linelist
- Issue #157: Create new parameters
min_lineage_sizeandmin_private_mutsto control lineage splitting intoX*-like.
Plot
- Issue #17: Create script to plot lineage assignment changes between versions using a Sankey diagram.
- Issue #82: Change epiweek start from Monday to Sunday.
- Issue #111: Fix breakpoint distribution axis that was empty for clade.
- Issue #152: Fix file saving bug when largest lineage has
/characters.
Report
- Issue #88: Add pipeline and nextclade versions to powerpoint slides as footer. This required adding
--summaryas param toreport.
Validate
- Issue #56: Change rule
validatefrom simply counting the number of positives to validating the fieldslineage,breakpoints,parents_clade. This involves adding a new default parameterexpectedfor rulevalidateindefaults/parameters.yaml.
Designated Lineages
- Issue #149:
XA - Issue #148:
XB - Issue #147:
XC - Issue #146:
XD - Issue #145:
XE - Issue #144:
XF - Issue #143:
XG - Issue #141:
XH - Issue #142:
XJ - Issue #140:
XK - Issue #139:
XL - Issue #138:
XM - Issue #137:
XN - Issue #136:
XP - Issue #135:
XQ - Issue #134:
XR - Issue #133:
XS - Issue #132:
XT - Issue #131:
XU - Issue #130:
XV - Issue #129:
XW - Issue #128:
XY - Issue #127:
XZ - Issue #126:
XAA - Issue #125:
XAB - Issue #124:
XAC - Issue #123:
XAD - [Issue #122](https://github.com...
v0.4.2 - Bugfix and Enhancement
v0.4.2
Notes
This is a minor bug fix and enhancement release with the following changes:
Linelist
- Issue #70: Fix missing
sc2rfversion fromrecombinant_classifier_dataset - Issue #74: Correctly identify
XN-likeandXP-like. Previously, these were just assignedXN/XPregardless of whether the estimated breakpoints conflicted with the curated ones. - Issue #76: Mark undesignated lineages with no matching sc2rf lineage as
unpublished.
Plot
- Issue #71: Only truncate
cluster_idwhile plotting, not in table generation. - Issue #72: For all plots, truncate the legend labels to a set number of characters. The exception to this are parent labels (clade,lineage) because the full label is informative.
- Issue #73, #75: For all plots except breakpoints, lineages will be defined by the column
recombinant_lineage_curated. Previously it was defined by the combination ofrecombinant_lineage_curatedandcluster_id, which made cluttered plots that were too difficult to interpret. - New parameter
--lineage-colwas added toscripts/plot_breakpoints.pyto have more control on whether we want to plot the raw lineage (lineage) or the curated lineage (recombinant_lineage_curated).
Commits
8953ef03docs: add CHANGELOG for v0.4.27ec5ccc6docs: add notes for v0.4.21b3b1f1dscript: restore column name to recombinant_classifer_dataset901caf98script: restore recombinant_lineage_curated of -like lineagesd6be9611script: change internal delim of classifier for #70cdb4a78ascript: fix recombinant_classifier missing sc2rf for #70bf7a4e57script: mark undesignated lineages with no matching sc2rf lineage as unpublished for #7646f6d754workflow: update linelists and plotting for #74 and #75c03dd3bescript: don't split largest by cluster id for #73e9802e79script: majority of plots will not split by cluster_id for #73bafb38fbscript: fix cluster ID truncation for issue #71ab712593resources: curate and test breakpoints for proposed895
v0.4.1 - Bugfix
v0.4.1
Notes
This is a minor bug fix release with the following changes:
- Issue #63: Remove
usherandprotobuffrom the conda environment. - Issue #68: Remove ncov as a submodule.
- Issue #69: Remove 22C and 22D from
sc2rf/mapping.csvandsc2rf/virus_properties.json, as these interfere with breakpoint detection for XAN.
Commits
v0.4.0 - BA.5 and UShER Removal
v0.4.0
Notes
General
v0.4.0 has been trained and validated on the latest generation of SARS-CoV-2 Omicron clades (ex. 22A/BA.4 and 22B/BA.5). Recombinant sequences involving BA.4 and BA.5 can now be detected, unlike in v0.3.0 where they were not included in the sc2rf models.
v0.4.0 is also a major update to how sequences are categorized into lineages/clusters. A recombinant lineage is now defined as a group of sequences with a unique combination of:
- Lineage assignment (ex.
XM) - Parental clades (ex.
Omicron/21K,Omicron/21L) - Breakpoints (ex.
17411:21617) - NEW: Parental lineages (ex.
BA.1.1,BA.2.12.1)
Novel recombinants (i.e. undesignated) can be identified by a lineage assignment that does not start with X* (ex. BA.1.1) or with a lineage assignment that contains -like (ex. XM-like). A cluster of sequences may be flagged as -like if one of the following criteria apply:
-
The lineage assignment by Nextclade conflicts with the published breakpoints for a designated lineage (
resources/breakpoints.tsv).- Ex. An
XEassigned sample has breakpoint11538:12879, which conflicts with the publishedXEbreakpoint (ex. 8394:12879). This will be renamedXE-like.
- Ex. An
-
The cluster has 10 or more sequences, which share at least 3 private mutations in common.
- Ex. A large cluster of sequences (N=50) are assigned
XM. However, these 50 samples share 5 private mutationsT2470C,C4586T,C9857T,C12085T,C26577Gwhich do not appear in trueXMsequences. These will be renamedXM-like. Upon further review of the reported matching pango-designation issues (460,757,781,472,798), we find this cluster to be a match toproposed798.
- Ex. A large cluster of sequences (N=50) are assigned
The ability to identify parental lineages and private mutations is largely due to improvements in the newly released nextclade datasets, , which have increased recombinant lineage accuracy. As novel recombinants can now be identified without the use of the custom UShER annotations (ex. proposed771), all UShER rules and output have been removed. This significantly improves runtime, and reduces the need to drop non-recombinant samples for performance. The result is more comparable output between different dataset sizes (4 samples vs. 400,000 samples).
Note! Default parameters have been updated! Please regenerate your profiles/builds with:
scripts/create_profile.sh --data data/custom
Datasets
- Issue #49: The tutorial lineages were changed from
XM,proposed467,miscBA1BA2Post17k, toXD,XH,XAN. The previous tutorial sequences had genome quality issues. - Issue #51: Add
XANto the controls dataset. This is BA.2/BA.5 recombinant. - Issue #62: Add
XAKto the controls dataset. This is BA.2/BA.1 VUM recombinant monitored by the ECDC.
Nextclade
- Issue #46:
nextcladeis now run twice. Once with the regularsars-cov-2dataset and once with thesars-cov-2-no-recombdataset. Thesars-cov-2-no-recombdataset is used to get the nucleotide substitutions before recombination occurred. These are identified by taking thesubstitutionscolumn, and excluding the substitutions found inprivateNucMutations.unlabeledSubstitutions. The pre-recombination substitutions allow us to identify the parental lineages by querying cov-spectrum. - Issue #48: Make the
exclude_cladescompletely optional. Otherwise an error would be raised if the user didn't specify any. - Issue #50: Upgrade from
v1.11.0tov2.3.0. Also upgrade the default dataset tags to 2022-07-26T12:00:00Z which had significant bug fixes. - Issue #51: Relax the recombinant criteria, by flagging sequences with ANY labelled private mutations as a potential recombinant for further downstream analysis. This was specifically for BA.5 recombinants (ex.
XAN) as no other columns from thenextcladeoutput indicated this could be a recombinant. - Restrict
nextcladeoutput tofasta,tsv(alignment and QC table). This saves on file storage, as the other default output is not used.
sc2rf
- Issue #51:
sc2rfis now run twice. First, to detect recombination between clades (ex.Delta/21J&Omicron/21K). Second, to detect recombination within Omicron (ex.Omicron/BA.2/21L&Omicron/BA.5/22B). It was not possible to define universal parameters forsc2rfthat worked for both distantly related clades, and the closely related Omicron lineages. - Issue #51: Rename parameter
cladestoprimary_cladesand add new parametersecondary_cladesfor detecting BA.5. - Issue #53: Identify the parental lineages by splitting up the observed mutations (from
nextclade) into regions by breakpoint. Then query the list of mutations in https://cov-spectrum.org and report the lineage with the highest prevalence. - Tested out
--enable-deletionsagain, which caused issues forXD. This confirms that using deletions is still ineffective for defining breakpoints. - Add
B.1.631andB.1.634tosc2rf/mapping.tsvand as potential clades in the default parameters. These are parents forXB. - Add
B.1.438.1tosc2rf/mapping.tsvand as a otential clade in the default parameters. This is a parent forproposed808. - Require a recombinant region to have at least one substitution unique to the parent (i.e. diagnostic). This reduces false positives.
- Remove the debugging mode, as it produced overly verbose output. It is more efficient to rerun manually with custom parameters tailored to the kind of debugging required.
- Change parent clade nomenclature from
Omicron/21Kto the more comprehensiveOmicron/BA.1/21K. This makes it clear which lineage is involved, since it's not always obvious how Nextclade clades map to pango lineages.
UShER
- Issue #63: All UShER rules and output have been removed. First, because the latest releases of nextclade datasets (tag
2022-07-26T12:00:00Z) have dramatically improved lineage assignment accuracy for recombinants. Second, was to improve runtime and simplicity of the workflow, as UShER adds significantly to runtime.
Linelist
- Issue #30: Fixed the bug where distinct recombinant lineages would occasionally be grouped into one
cluster_id. This is due to the new definition for recombinant lineages (see General) section, which now includes parental lineages and have sufficient resolving power. - Issue #46: Added new column
parents_subs, which are the substitutions found in the parental lineages before recombination occurred using thesars-cov-2-no-recombnextclade dataset. Also added new columns:parents_lineage,parents_lineage_confidence, based on queryingcov-spectrumfor the substitutions found inparents_subs. - Issue #53: Added new column
cov-spectrum_querywhich includes the substitutions that are shared by ALL sequences of the recombinant lineage. - Added new column
cluster_privateswhich includes the private substitutions shared by ALL sequences of the recombinant lineage. - Renamed
parentscolumn toparents_clade, to differentiate it from the new columnparents_lineage.
Plot
- Issue #4, Issue #57: Plot distributions of each parent separately, rather than stacking on one axis. Also plot the substitutions as ticks on the breakpoints figure.
| v0.3.0 | v0.4.0 |
|---|---|
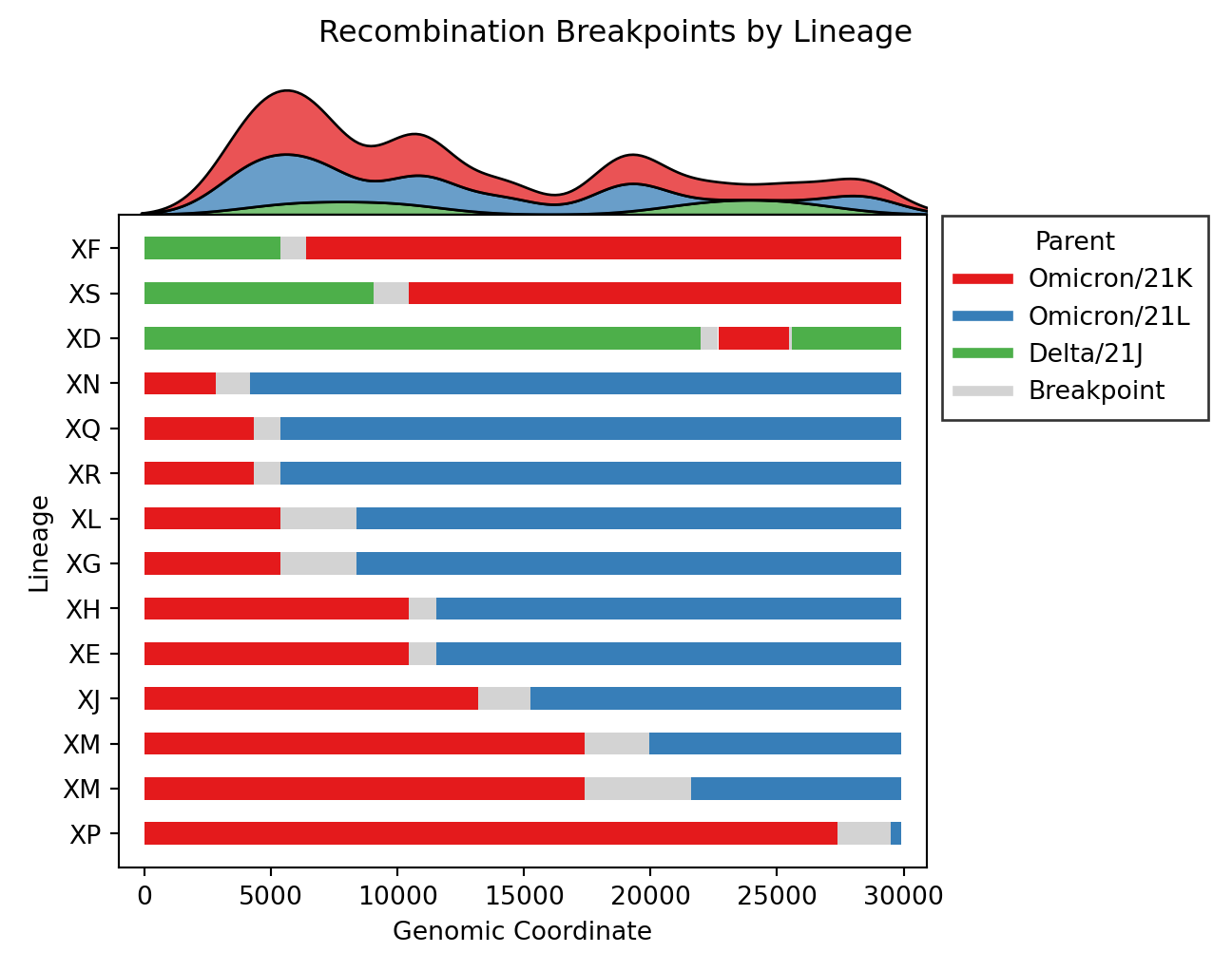 |
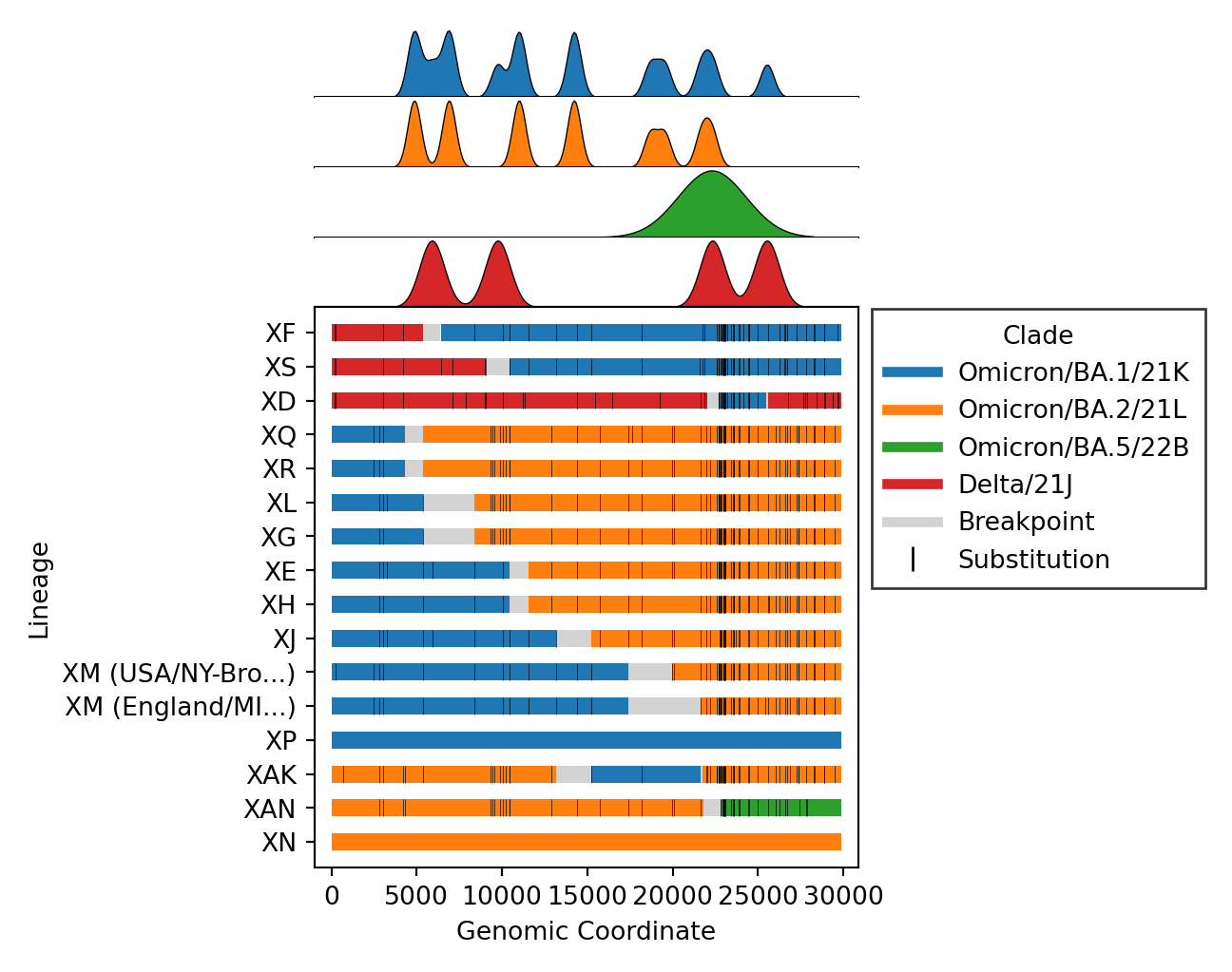 |
- Issue #46: Plot breakpoints separately by clade and lineage. In addition, distinct clusters within the same recombinant lineage are noted by including their cluster ID as a suffix. As an example, please see
XM (USA) and X (England)below. Where the lineage is the same (XM), but the breakpoints differ, as do the parental lineages (BA.2vsBA.2.12.1). These clusters are distinct becauseXM (England)lacks substitutions occurring around position 20000.
| ...
v0.3.0 - No Recombinants, No Problems
v0.3.0
Notes
Major Changes
-
By default, all sequences will go through all steps of the pipeline. This prevents pipeline errors when no recombinant sequences are detected. See the FAQ for info on changing this setting.
-
Default parameters have been updated! Please regenerate your profiles/builds with:
scripts/create_profile.sh --data data/custom
-
Rule outputs are now in sub-directories for a cleaner
resultsdirectory. -
The in-text report (
report.pptx) statistics are no longer cumulative counts of all sequences. Instead they, will match the reporting period in the accompanying plots.
Bug Fixes
- Improve subtree collapse effiency (#35).
- Improve subtree aesthetics and filters (#20).
- Fix issues rendering as float (#29).
- Explicitly control the dimensions of plots for powerpoint embedding.
- Remove hard-coded
extra_cols(#26). - Fix mismatch in lineages plot and description (#21).
- Downstream steps no longer fail if there are no recombinant sequences (#7).
Output
-
Output new
_historicalplots and slides for plotting all data over time. -
Output new file
parents.tsvto summarize recombinant sequences by parent. -
Order the colors/legend of the stacked bar
plotsby number of sequences. -
Include lineage and cluster id in filepaths of largest plots and tables.
-
Rename the linelist output:
linelist.tsvpositives.tsvnegatives.tsvfalse_positives.tsvlineages.tsvparents.tsv
-
The
report.xlsxnow includes the following tables:- lineages
- parents
- linelist
- positives
- negatives
- false_positives
- summary
- issues
Data
-
Create new controls datasets:
controls-negativescontrols-positivescontrols
-
Add versions to
genbank_accessionsforcontrols.
Programs
- Upgrade UShER to v0.5.4 (possibly this was done in a prior ver).
- Remove
taxoniumandchronumentalfrom the conda env.
Parameters
-
Add parameters to control whether negatives and false_positives should be excluded:
exclude_negatives: falsefalse_positives: false
-
Add new optional param
max_placementsto rulelinelist. -
Remove
--show-private-mutationsfromdebug_argsof rulesc2rf. -
Add optional param
--sc2rf-dirtosc2rfto enable execution outside ofsc2rfdir. -
Add params
--output-csvand--output-ansito the wrapperscripts/sc2rf.sh. -
Remove params
nextclade_refandcustom_reffrom rulenextclade. -
Change
--breakpoints 0-10insc2rf.
Workflow
- Add new rule
usher_columnsto augment the base usher metadata. - Add new script
parents.py, plots, and report slide to summarize recombinant sequences by parent. - Make rules
plotandreportmore dynamic with regards to plots creation. - Exclude the reference genome from alignment until
faToVcf. - Include the log path and expected outputs in the message for each rule.
- Use sub-functions to better control optional parameters.
- Make sure all rules write to a log if possible (#34).
- Convert all rule inputs to snakemake rule variables.
- Create and document a
create_profile.shscript. - Implement the
--low-memorymode parameter within the scriptusher_metadata.sh.
Continuous Integration
-
Re-rename tutorial action to pipeline, and add different jobs for different profiles:
- Tutorial
- Controls (Positive)
- Controls (Negative)
- Controls (All)
Pull Requests
pull/40v0.3.0 stability update part 2pull/8Add XS and XQ to controls.pull/19docs: add lenaschimmel as a contributor for codepull/12Tutorial dataset and map panel for Auspice subtreespull/11Add a tutorial profilepull/14Plots and PowerPointspull/15New rule: parentspull/39v0.3.0 stability update
Commits
2f8b498adocs: update changelog for v0.3.00486d3bedocs: add updating section to readme for issue #33e8eda400resources: updates issues with curate breakpoints12e3700fbug: catch empty dataframe in plotd1ccca2aworkflow: first successful high-throughput runcd741a10workflow: add new rules plot_historical and report_historicalc2cc1380env: remove openpyxl from environment7dc7c039workflow: remove rule report_redact #319ca5f822script: rearrange merge file order in summaryaa28eb9fworkflow: create new rule report_redact for #314748815denv: add openpyxl to environment for excel parsing in python0060904ascript: template duplicate labelling in usher_collapse for latera82359a7data: add accession versions to controls metadataaf7341aadata: add accession versions to controls metadatad860a4c8workflow: add new rule usher_columns to augment the base usher metadata2511673dimprove subtree collapse effiency (#35) and output aesthetics (#20)1e81be3bbug: remove non-existant param --log in rule usher_metadata02198b4cscript: add logging to usher_collapsed40d3d78ci: don't run pipeline just for images changesb880d9c8docs: update powerpoint image to proper ver- See CHANGELOG.md for additional commits.
v0.2.1 - Plots and Powerpoints
v0.2.1
Notes
Params
- New optional param
motifsfor rulesc2rf_recombinants. - New param
weeksfor new ruleplot. - Removed
prev_linelistparam.
Output
- Switch from a pdf
reportto powerpoint slides for better automation. - Create summary plots.
- Split
reportrule intolinelistandreport. - Output
svgplots.
Workflow
- New rule
plot. - Changed growth calculation from a comparison to the previous week to a score of sequences per day.
- Assign a
cluster_idaccording to the first sequence observed in the recombinant lineage. - Define a recombinant lineage as a group of sequences that share the same:
- Lineage assignment
- Parents
- Breakpoints or phylogenetic placement (subtree)
- For some sequences, the breakpoints are inaccurate and shifted slightly due to ambiguous bases. These sequences can be assigned to their corresponding cluster because they belong to the same subtree.
- For some lineages, global prevalence has exceeded 500 sequences (which is the subtree size used). Sequences of these lineages are split into different subtrees. However, they can be assigned to the correct cluster/lineage, because they have the same breakpoints.
- Confirmed not to use deletions define recombinants and breakpoints (differs from published)?
Programs
- Move
sc2rf_recombinants.pytopostprocess.pyin ktmeaton fork ofsc2rf. - Add false positives filtering to
sc2rf_recombinantsbased on parents and breakpoints.
Docs
- Add section
ConfigurationtoREADME.md.
Pull Requests
pull/14Plots and PowerPoints
Commits
c2369c75update CHANGELOG after README overhaul9c8a774eupdate autologs to exclude first blank line in notes2a8a7af5overhaul README9c2bd2f5change asterisks to dashes46d4ec81update autologs to allow more complex notes contenta01a903csplit docs into dev and todo23e8d715change color palette for plotting785b8a19add optional param motifs for sc2rf_recombinantsd1c1559erestore pptx template to regular view6adc5d32add seaborn to environment35a04471add changelog to report pptx99e98aa7add epiweeks to environment1644b1fcadd pptx report1ab93aff(broken) start plotting094530f0swithc sc2rf to a postprocess script02193d6etry generalizing sc2rf post-processing