{kind=link}
{kind=link}
{kind=link}
![]()
When publishing results obtained with DFT-QE WaNo, please consider citing it.
Let's start exploring your materials' properties with ease and precision! Welcome to the DFT-QE WaNo, which performs DFT calculations using the powerful and flexible Quantum Espresso code! Quantum Espresso is an open-source software package that can handle many systems, from small molecules to extended solids. It provides a wide range of exchange-correlation functionals. It can be used to perform various types of calculations, including ground state properties, electronic structure, and phonon calculations. With our WaNo, the pseudopotentials (SSSP Efficiency (version 1.1.2)) are automatically identified based on the chemical species specification. All input files are generated or loaded automatically after reading the geometry file in .cif format, making it easy for you to start.
To get this WaNo up and running on your available computational resources, make sure to have the below libraries installed on Python 3.6 or newer.
1. Atomic Simulation Environment (ASE).
2. Python Materials Genomics (Pymatgen).
3. Numpy, os, sys, re, yaml.
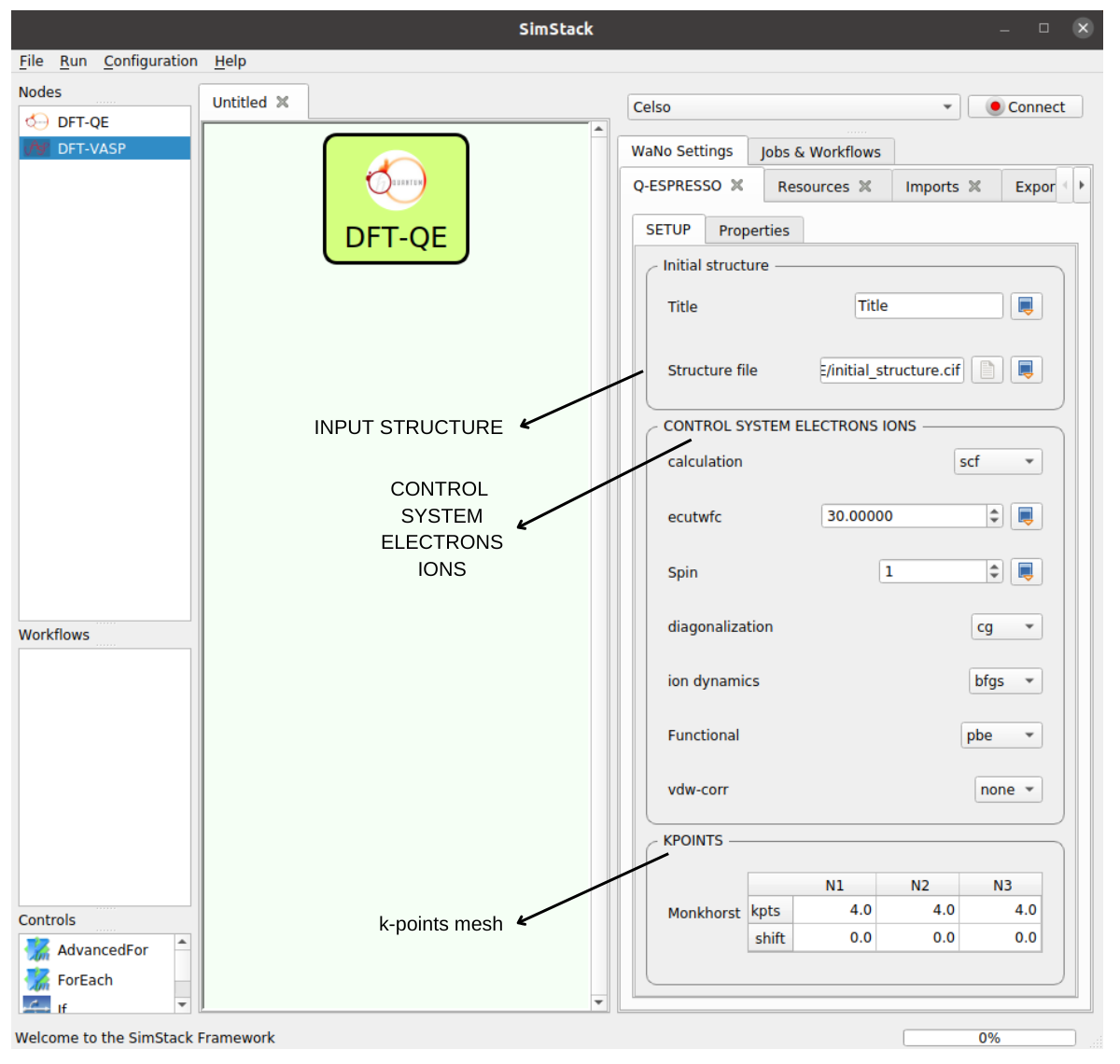
- SETUP tab: See the GUI of this WaNo. as an option. We can set the initial structure, Controls system, and KPOINTS.
- Properties tab: (:warning: The
qe_results.ymldatabase is replacing this tab. Check down below to see the available properties in the database!).
QEIN.out- This file contains vital information about the simulation of the system.
Title.save- A folder containing the charge-density.dat, paw.txt, pseudo potentials, and all wfcdw
.datfiles.
- A folder containing the charge-density.dat, paw.txt, pseudo potentials, and all wfcdw
qe_results.yml- This database comprises all inputs in the
rendered_wano.ymlfile as well a set of keys and values of the following properties: total_energy, potential_energy, kinetic_energy, cell, cell_lengths_and_angles, positions, forces, chemical_formula, chemical_symbols, center_of_mass, volume, temperature, all_distances, masses, atomic_numbers, global_number_of_atoms, initial_charges, band_gap, convergence, datetime, and user.
- This database comprises all inputs in the
- Step 1. Move the DFT-QE folder to the WaNo directory.
- Step 2. Open Simstack on your compute and connect to your remote resource.
- Step 3. Drag the WaNo from the top left menu to the SimStack canvas as shown in Fig 1.
- Step 4. A double click on the WaNo will allow you to make the setups in the Input parameters.
- Step 5. Name your WaNo with
Ctrl+S, and running it withCtrl+Rcommand.
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 957189. The project is part of BATTERY 2030+, the large-scale European research initiative for inventing the sustainable batteries of the future.
Developer: Celso Ricardo C. Rêgo, Multiscale Materials Modelling and Virtual Design, Institute of Nanotechnology, Karlsruhe Institute of Technology https://www.int.kit.edu/wenzel.php
Licensed under the KIT License.