-- Yang Eric Li
In the last chapter, we use two strategy to align the reads to the genome and different RNA types. As we extract the sequence for each RNA type and mapped reads to them, thus, the coordinates for each reads stored in bam file is not the position of where they aligned to the genome, but the transcripts for each RNA types. We use RSEM ( https://github.com/deweylab/RSEM) to solve these problem.
# build bowtie2 index using RSEM
rsem-prepare-reference --gtf /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gtf --bowtie2 /BioII/lulab_b/shared/genomes/human_hg38/sequence/GRCh38.p12.genome.fa miRNA.indexDir/
# align reads to miRNA
bowtie2 -p 4 --sensitive-local --no-unal --un NC_1.unAligned.fq -x miRNA.indexDir/ NC_1.rRNA_exon.unmapped.fastq -S NC_1.miRNA.sam
# convert the coordinates in bam files
rsem-tbam2gbam miRNA.indexDir/ NC_1.miRNA.sam NC_1.miRNA.rsem.bam
# sort bam file
samtools sort NC_1.miRNA.rsem.bam > NC_1.miRNA.sorted.bam
# build bam index
samtools index NC_1.miRNA.sorted.bam
# create bedGraph
bedtools genomecov -ibam NC_1.miRNA.sorted.bam -bga -split -scale 1.0 | sort -k1,1 -k2,2n > NC_1.miRNA.sorted.bedGraph
# convert bedGraph to bigWig
bedGraphToBigWig NC_1.miRNA.sorted.bedGraph /BioII/lulab_b/shared/genomes/human_hg38/sequence/hg38.chrom.sizes NC_1.miRNA.sorted.bw
Note http://bedtools.readthedocs.io/en/latest/content/tools/genomecov.html
- 1.Make tag directories for each experiment
#works with sam or bam (samtools must be installed for bam)
makeTagDirectory NC_1.miRNA.tagsDir/ NC_1.miRNA.sorted.bam
If the experiment is strand specific paired end sequencing, add "-sspe" to the end. If it's unstranded paired-end sequencing, no extra options are needed. makeTagDirectory tags_Dir/ inputfile.sam -format sam -sspe
- 2.Make bedGraph visualization files for each tag directory
# Add "-strand separate" for strand-specific sequencing
makeUCSCfile NC_1.miRNA.tagsDir/ -fragLength given -o auto
(repeat for other tag directories)

in our case, try to load the bam and bigwig format file


- alignment: bam/sam format
convert sam to bam
samtools view -S -b NC1.miRNA_hg38_mapped.sam > NC1.miRNA_hg38_mapped.bam
- annotaion: gtf/gff/gff3 format
convert gff to gtf
gffread tRNA.gff -T -o tRNA.gtf
Given a file with aligned sequencing reads(.sam/.bam) and a list of genomic features(.gtf), a common task is to count how many reads map to each feature(gene).
Usage
htseq-count [options] <alignment_files> <gff_file>
in our case
htseq-count -m intersection-strict --idattr=Name --type=miRNA_primary_transcript NC_1.miRNA_hg38_mapped.sam /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gff > NC_1.miRNA.htseq.counts
-
3 overlap resolution models
*
-
We shared our snakemake package used for exRNA-seq expression matrix construction.Github.
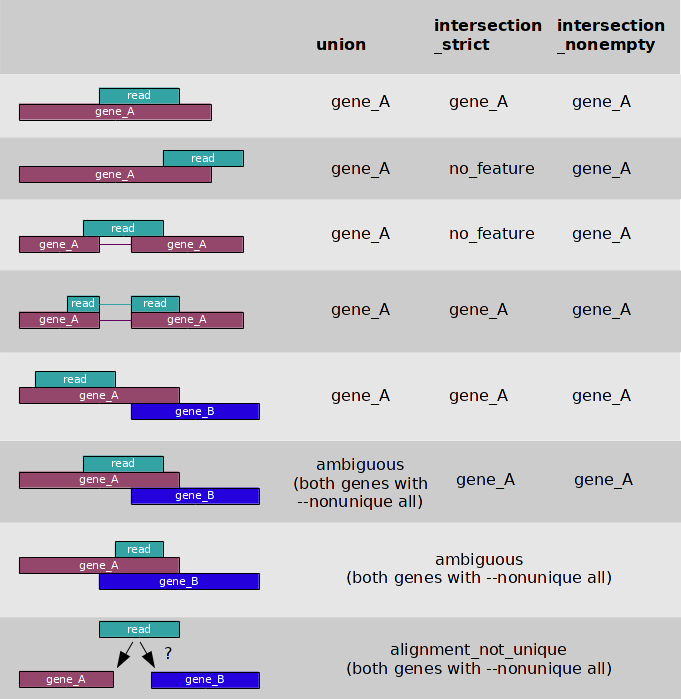
Tips
- --nonunique
- --nonunique none (default): the read (or read pair) is counted as ambiguous and not counted for any features. Also, if the read (or read pair) aligns to more than one location in the reference, it is scored as alignment_not_unique.
- --nonunique all: the read (or read pair) is counted as ambiguous and is also counted in all features to which it was assigned. Also, if the read (or read pair) aligns to more than one location in the reference, it is scored as alignment_not_unique and also separately for each location.
- Notice that when using --nonunique all the sum of all counts will not be equal to the number of reads (or read pairs), because those with multiple alignments or overlaps get scored multiple times.
Notes
- -m/--mode {mode}
- --nonunique={none/all}
- -s/--stranded {yes/no/reverse}.
- -a {minaqual}.
- -t/--type {feature type}. (defult: exon)
- -i/--idattr {id attribute}, GFF attribute to be used as feature ID. (defult: gene_id)
Usage
Summarize a BAM format dataset:
featureCounts -t exon -g gene_id -a annotation.gtf -o counts.txt mapping_results_SE.bam
Summarize multiple datasets at the same time:
featureCounts -t exon -g gene_id -a annotation.gtf -o counts.txt library1.bam library2.bam library3.bam
in our case
featureCounts -t miRNA_primary_transcript -g Name -a /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gff -o NC_1.miRNA.featureCounts.counts NC_1.miRNA.sorted.bam
Tips By default, featureCounts does not count reads overlapping with more than one feature. Users can use the -O option to instruct featureCounts to count such reads (they will be assigned to all their overlapping features or meta-features).
- RPKM: Reads Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的reads)
- FPKM: Fragments Per Kilobase of exon model per Million mapped fragments(每千个碱基的转录每百万映射读取的fragments, 对于Pair-end sequencing, two paired reads should be mapped simultaneously)
RPKM = total exon reads/ (mapped reads (Millions) * exon length(KB))
- RPM/CPM: Reads/Counts of exon model per Million mapped reads (每百万映射读取的reads)
RPM=total exon reads / mapped reads (Millions)
Usage
- 0.Align FASTQ reads using STAR or similar 'splicing aware' genome alignment algorithm
- 1.Make tag directories for each experiment
#works with sam or bam (samtools must be installed for bam)
makeTagDirectory Exp1r1/ inputfile1r1.sam -format sam
makeTagDirectory Exp1r2/ inputfile1r2.sam -format sam
If the experiment is strand specific paired end sequencing, add "-sspe" to the end. If it's unstranded paired-end sequencing, no extra options are needed. makeTagDirectory Exp1/ inputfile.sam -format sam -sspe
- 2. Quantify gene expression across all experiments for clustering and reporting (-rpkm / -rpm / -log2 / -quantile / -sqrt):
# May also wish to use "-condenseGenes" if you don't want multiple isoforms per gene
analyzeRepeats.pl rna hg38 -strand both -count exons -d Exp1r1/ Exp1r2 Exp2r1/ Exp2r2/ -rpkm > rpkm.txt
# Use this result for gene expression clustering, PCA, etc.
- 3. Quantify gene expression as integer counts for differential expression (-noadj)
# May also wish to use "-condenseGenes" if you don't want multiple isoforms per gene
analyzeRepeats.pl rna hg38 -strand both -count exons -d Exp1r1/ Exp1r2 Exp2r1/ Exp2r2/ -noadj > raw.txt
in our case
makeTagDirectory NC_1.miRNA.tagsDir/ NC_1.miRNA.sorted.bam
# raw counts
analyzeRepeats.pl /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gtf hg38 -count exons -d NC_1.miRNA.tagsDir/ -noadj > NC_1.miRNA.homer.counts
# calculate rpkm
analyzeRepeats.pl /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gtf hg38 -count exons -d NC_1.miRNA.tagsDir/ -rpkm > NC_1.miRNA.homer.rpkm
# calculate rpm/cpm
analyzeRepeats.pl /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gtf hg38 -count exons -d NC_1.miRNA.tagsDir/ -norm 1e7 > NC_1.miRNA.homer.rpm
Tips: in default, homer do not use gff format file but gtf format. look into the difference between:
/BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gtf /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gff
TPM:Transcripts Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的Transcripts)
TPMi=(Ni/Li)*1000000/sum(Ni/Li+……..+ Nm/Lm)
Merge expression matrix of different RNA types together, then convert RPKM to TPM.
cat NC_1.miRNA.homer.rpkm NC_1.piRNA.homer.rpkm NC_1.Y_RNA_exon.homer.rpkm NC_1.snRNA.homer.rpkm NC_1.srpRNA_exon.homer.rpkm NC_1.tRNA.homer.rpkm NC_1.lncRNA_exon.homer.rpkm NC_1.mRNA_exon.homer.rpkm | grep -v "Transcript" | cut -f 1-6,9 | sed -e "1i geneID\tchr\tstart\tend\tstrand\tLength\tRPKM" > NC_1.merge.homer.rpkm.tsv
R script:
# Convert RPKM to transcripts per million (TPM).
> mx <- read.table("NC_1.merge.homer.rpkm.tsv",head=T,sep="\t")
> mx$TPM <- mx$RPKM/sum(mx$RPKM)*10^6
> head(mx)
geneID chr start end strand Length RPKM TPM
1 MI0014230 chr18 14830166 14830242 + 76 0.000 0.0000000
2 MI0017288 chr8 9048445 9048518 + 73 5.675 0.3932538
3 MI0022961 chr3 123161794 123161879 + 85 14.622 1.0132436
4 MI0003616 chr10 24275685 24275781 + 96 0.000 0.0000000
5 MI0017866 chr2 75090812 75090914 + 102 4.062 0.2814797
6 MI0000762 chrX 50008964 50009028 + 64 139.173 9.6441082
> mx <- read.table("foo",head=T,sep="\t")
> head(mx)
geneID chr start end strand Length RPKM
1 MI0014230 chr18 14830166 14830242 + 76 0.000
2 MI0017288 chr8 9048445 9048518 + 73 5.675
3 MI0022961 chr3 123161794 123161879 + 85 14.622
4 MI0003616 chr10 24275685 24275781 + 96 0.000
5 MI0017866 chr2 75090812 75090914 + 102 4.062
6 MI0000762 chrX 50008964 50009028 + 64 139.173
> mx$TPM <- mx$RPKM/sum(mx$RPKM)*10^6
> head(mx)
geneID chr start end strand Length RPKM TPM
1 MI0014230 chr18 14830166 14830242 + 76 0.000 0.0000000
2 MI0017288 chr8 9048445 9048518 + 73 5.675 0.3932538
3 MI0022961 chr3 123161794 123161879 + 85 14.622 1.0132436
4 MI0003616 chr10 24275685 24275781 + 96 0.000 0.0000000
5 MI0017866 chr2 75090812 75090914 + 102 4.062 0.2814797
6 MI0000762 chrX 50008964 50009028 + 64 139.173 9.6441082
use linux command (cut, awk, sed, paste...) generate expression matrix
use homer:
analyzeRepeats.pl /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gtf hg38 \
-d NC_1.miRNA.tagsDir/ NC_2.miRNA.tagsDir/ NC_3.miRNA.tagsDir/ \
BeforeSurgery_1.miRNA.tagsDir/ BeforeSurgery_2.miRNA.tagsDir/ BeforeSurgery_3.miRNA.tagsDir/ \
AfterSurgery_1.miRNA.tagsDir/ AfterSurgery_2.miRNA.tagsDir/ AfterSurgery_3.miRNA.tagsDir/ \
-noadj > hcc_example.miRNA.homer.ct.tsv
cut -f 1,9- $path0/04.counts/hcc_example.homer.ct.tsv $path0/04.counts/hcc_example.homer.ct.mx
# manually modify the header
# geneID NC_1 NC_2 NC_3 BeforeSurgery_1 BeforeSurgery_2 BeforeSurgery_3 AfterSurgery_1 AfterSurgery_2 AfterSurgery_3
# hsa-mir-1295a 0.000 1.000 0.000 6.000 11.000 5.000 1.000 18.000 3.000
# hsa-mir-1248 6.000 15.000 5.000 49.000 4.000 63.000 118.000 22.000 81.000
# hsa-mir-130b 127.000 118.000 146.000 148.000 336.000 167.000 92.000 129.000 352.000
# hsa-mir-3142 1.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000
# hsa-mir-541 2.000 0.000 4.000 1.000 21.000 0.000 0.000 0.000 2.000
# R script
> mx <- read.table("hcc_example.miRNA.homer.ct.mx",sep="\t",header=T)
> head(mx[,1:5])
geneID NC_1 NC_2 NC_3 BeforeSurgery_1
1 hsa-mir-1295a 0 1 0 6
2 hsa-mir-1248 6 15 5 49
3 hsa-mir-130b 127 118 146 148
4 hsa-mir-3142 1 0 0 0
5 hsa-mir-541 2 0 4 1
6 hsa-mir-3130-2 5 7 11 2
> colSums(mx[,2:ncol(mx)])
NC_1 NC_2 NC_3 BeforeSurgery_1 BeforeSurgery_2
3822770 3113065 4155202 7187702 5865615
BeforeSurgery_3 AfterSurgery_1 AfterSurgery_2 AfterSurgery_3
5015987 4455862 7848293 3167270
> apply(mx[,2:ncol(mx)], 2, function(c)sum(c!=0))
NC_1 NC_2 NC_3 BeforeSurgery_1 BeforeSurgery_2
883 868 881 865 989
BeforeSurgery_3 AfterSurgery_1 AfterSurgery_2 AfterSurgery_3
910 848 915 951
Purpose Filtering low-expression genes improved DEG detection sensitivity.
Methods
- Filter the genes with a total counts < = threshold;
- Filter genes with maximum normalized counts across all samples < = threshold;
- Filter genes with mean normalized counts across all samples < = threshold.
Criteria Retain genes: (Reads >= 1 counts) >= 20% samples.
> filter_genes <- apply(
+ mx[,2:ncol(mx)],
+ 1,
+ function(x) length(x[x > 2]) >= 2
+ )
> mx_filterGenes <- mx[filter_genes,]
> cor.test(mx_filterGenes$NC_1,mx_filterGenes$NC_2,methods="spearman")
> pairs(~NC_1+NC_2+NC_3,data=mx_filterGenes)
> dev.off()

-
build bedGraph/bigWig for visualization
## 1.1 use BEDtools and UCSC Kent Utilities samtools sort NC_1.miRNA.rsem.bam > NC_1.miRNA.sorted.bam samtools index NC_1.miRNA.sorted.bam bedtools genomecov -ibam NC_1.miRNA.sorted.bam -bga -split -scale 1.0 | sort -k1,1 -k2,2n > NC_1.miRNA.sorted.bedGraph bedGraphToBigWig NC_1.miRNA.sorted.bedGraph /BioII/lulab_b/shared/genomes/human_hg38/sequence/hg38.chrom.sizes NC_1.miRNA.sorted.bw ## 1.2 use homer makeTagDirectory NC_1.miRNA.tagsDir/ NC_1.miRNA.sorted.bam makeUCSCfile NC_1.miRNA.tagsDir/ -fragLength given -o auto -
count for expression value
# using HTSeq samtools view NC_1.miRNA.rsem.bam > NC_1.miRNA.rsem.sam htseq-count -m intersection-strict --idattr=Name --type=miRNA_primary_transcript NC_1.miRNA.rsem.sam /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gff > NC_1.miRNA.htseq.counts # using featureCounts featureCounts -t miRNA_primary_transcript -g Name -a /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gff -o NC_1.miRNA.featureCounts.counts NC_1.miRNA.sorted.bam # using homer analyzeRepeats.pl /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gtf hg38 -count exons -d NC_1.miRNA.tagsDir/ -noadj > NC_1.miRNA.homer.countsa -
generate expression matrix
# use homer analyzeRepeats.pl /BioII/lulab_b/shared/genomes/human_hg38/gtf/miRNA.gtf hg38 \ -d NC_1.miRNA.tagsDir/ NC_2.miRNA.tagsDir/ NC_3.miRNA.tagsDir/ \ BeforeSurgery_1.miRNA.tagsDir/ BeforeSurgery_2.miRNA.tagsDir/ BeforeSurgery_3.miRNA.tagsDir/ \ AfterSurgery_1.miRNA.tagsDir/ AfterSurgery_2.miRNA.tagsDir/ AfterSurgery_3.miRNA.tagsDir/ \ -noadj > hcc_example.miRNA.homer.ct.tsv -
cleaning the expression matrix
# R script
> mx <- read.table("hcc_example.miRNA.homer.ct.mx",sep="\t",header=T)
> head(mx[,1:5])
geneID NC_1 NC_2 NC_3 BeforeSurgery_1
1 hsa-mir-1295a 0 1 0 6
2 hsa-mir-1248 6 15 5 49
3 hsa-mir-130b 127 118 146 148
4 hsa-mir-3142 1 0 0 0
5 hsa-mir-541 2 0 4 1
6 hsa-mir-3130-2 5 7 11 2
# check library size (total mapped reads)
> colSums(mx[,2:ncol(mx)])
NC_1 NC_2 NC_3 BeforeSurgery_1 BeforeSurgery_2
3822770 3113065 4155202 7187702 5865615
BeforeSurgery_3 AfterSurgery_1 AfterSurgery_2 AfterSurgery_3
5015987 4455862 7848293 3167270
# check the number of detected genes
> apply(mx[,2:ncol(mx)], 2, function(c)sum(c!=0))
NC_1 NC_2 NC_3 BeforeSurgery_1 BeforeSurgery_2
883 868 881 865 989
BeforeSurgery_3 AfterSurgery_1 AfterSurgery_2 AfterSurgery_3
910 848 915 951
# filter genes
> filter_genes <- apply(
+ mx[,2:ncol(mx)],
+ 1,
+ function(x) length(x[x > 2]) >= 2
+ )
> mx_filterGenes <- mx[filter_genes,]
> head(mx_filterGenes[,1:5])
geneID NC_1 NC_2 NC_3 BeforeSurgery_1
1 hsa-mir-1295a 0 1 0 6
2 hsa-mir-1248 6 15 5 49
3 hsa-mir-130b 127 118 146 148
5 hsa-mir-541 2 0 4 1
6 hsa-mir-3130-2 5 7 11 2
7 hsa-mir-369 3506 1117 2086 12
# check the correlations between each samples
> cor.test(mx_filterGenes$NC_1,mx_filterGenes$NC_2,methods="spearman")
Pearson's product-moment correlation
data: mx_filterGenes$NC_1 and mx_filterGenes$NC_2
t = 33.508, df = 863, p-value < 2.2e-16
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.7214444 0.7795271
sample estimates:
cor
0.7519414
> pairs(~NC_1+NC_2+NC_3,data=mx_filterGenes)
> dev.off()
# save the results
write.table(mx_filterGenes,"hcc_example.miRNA.homer.ct.filtered.mx",sep="\t",quote=F,col.names=T,row.names=F)
https://github.com/lulab/training/tree/master/proj_exRNA/example_small/constructExpMx
cnode: /BioII/lulab_b/shared/shared_scripts/bioinfoTraining/constructExpMx
Level I:
-
learn how to construct the expression matrix.
-
compare the difference between HTSeq, featureCounts and homer
-
check the bam/bigWig file using IGV
-
QC and cleaning the expression matrix
Level II:
reference scripts: cnode: /home/younglee/projects/hcc_example/bin
-
convert bam/sam files coordinates to genome coordinates (for sequential mapping)
-
construct expression matrix for all RNA types (miRNA, piRNA, mRNA, lncRNA...)
-
QC and cleaning the expression matrix
Level III:
quantify miRNA using bam files from common (map to hg38 genome directly) and sequential mapping (map to multiple RNA types), respectively.
Compare the differences and explain why. (using RSEM: https://github.com/deweylab/RSEM, please check rsem-prepare-reference and rsem-tbam2gbam functions)